
咪唑甘油磷酸酯脱水酶与含氮杂环磷酸酯类抑制剂作用方式的分子模拟
2011-11-30 10:50杜凤沛姚广伟方萌萌
物理化学学报 2011年8期
申 涛 杜凤沛 刘 婷 姚广伟 吴 峥 方萌萌
徐筱杰2 路慧哲1,*
(1中国农业大学理学院,北京100193;2北京大学化学与分子工程学院,北京100871)
咪唑甘油磷酸酯脱水酶与含氮杂环磷酸酯类抑制剂作用方式的分子模拟
申 涛1杜凤沛1刘 婷1姚广伟1吴 峥1方萌萌1
徐筱杰2路慧哲1,*
(1中国农业大学理学院,北京100193;2北京大学化学与分子工程学院,北京100871)
国际上依据咪唑甘油磷酸酯脱水酶(IGPD)底物结构筛选,成功获得了一系列含氮杂环磷酸酯类化合物作为IGPD抑制剂,然而IGPD与含氮杂环磷酸酯类抑制剂间的作用模式尚不清楚.本研究利用Gaussian 03程序,基于密度泛函理论B3LYP方法,选择6-31G**基组优化含氮杂环磷酸酯类化合物,在确定其稳定构象的基础上利用分子对接、力学优化构建IGPD与其含氮杂环磷酸酯类抑制剂相互作用的复合物结构,基于化合物的电子结构(前线轨道能级及组成、原子电荷、自然键轨道等)、复合物的空间结构(抑制剂识别IGPD的功能域、分子间氢键、van der Waals相互作用等)探讨了IGPD与含氮杂环磷酸酯类抑制剂作用方式,确定了含氮杂环电荷分布、磷酸根离子电荷分布、前线轨道LUMO能级是影响抑制剂活性的内在因素,为进一步筛选、优化高效的新型除草剂提供了重要信息.
咪唑甘油磷酸酯脱水酶;抑制剂;分子对接;力学优化;密度泛函理论;前线轨道
1 引言
作为植物和微生物体内组氨酸生物合成的关键酶——咪唑甘油磷酸酯脱水酶(IGPD)已成为目前日益受到关注的除草剂潜在靶标,通过抑制酶的生物学活性即阻断组氨酸的合成可致植物死亡而达到除草的目的.IGPD位于组氨酸生物合成反应的第七步.由咪唑甘油磷酸酯(IGP)经脱水反应生成咪唑丙酮醇磷酸酯(IAP),可能的催化机理如图所1示,1该酶是植物和微生物体内组氨酸生物合成的关键酶之一.大量的研究表明,在植物和微生物体内,组氨酸的生物合成是至关重要的,而哺乳动物体内却不具备这种氨基酸的生物合成能力,即体内并无此酶.因此通过抑制IGPD的活性从而阻断杂草体内组氨酸的生物合成,可使杂草死亡而对人畜无害.1随着Tada等2进一步确认IGPD位于植物细胞叶绿体内,特别是Sinha等3报道IGPD催化亚基及其三聚体X射线晶体衍射结果后,IGPD成为近年来研究除草剂的潜在靶标酶.
Mori等4-6以IGPD的底物IGP(图1)为结构基础,通过生物合理设计的方法确认了以IGPD为靶标的一系列先导化合物,并测定了其IC50(图2).
一系列的生物学实验表明,以IGP为结构基础获得的IGPD抑制剂具有类似的母核结构:(1)含有氮原子的杂环;(2)邻位羟基;(3)磷酸根离子,然而其IC50值偏差很大.5本研究中利用上述抑制剂结构,通过密度泛函理论,选择6-31G**基组优化其构象,并通过振动频率计算确定其稳定结构.基于IGPD晶体结构,通过分子对接、力学优化搭建稳定的抑制剂与IGPD相互作用的复合物结构,通过分析抑制剂的电子结构以及复合物集合结构特点,从宏观和微观的角度探讨了基于IGP结构设计的一系列IGPD抑制剂与IGPD作用的可能模式,确定了抑制剂核心区域的结构特征,为进一步的IGPD抑制剂的筛选及优化提供了理论依据.
2 计算方法
2.1 量子化学计算

图1 IGPD可能的催化机理Fig.1 Proposed catalytic mechanism of IGPD1IGPD:imidazole glycerol phosphate(IGP)dehydratase;B:XHHXE,potential binding motif in IGPD

图2 基于IGP结构设计筛选的一系列先导化合物4-6Fig.2 Aseries of designed lead compounds based on IGPstructure4-6
IGPD抑制剂分子a-g(图2)及IGP的结构(图1)均采用Gaussian 03程序包7进行如下次序的优化:所有分子的几何构型先采用Hartree-Fock方法在3-21G基组水平条件下进行初步优化,在预优化的基础上采用密度泛函理论中B3LYP方法8在6-31G**基组9,10水平下对抑制剂进行分子构型的全优化计算,并通过振动频率的计算对其热稳定性进行了评价.计算的收敛精度采用Gaussian 03程序的内定值,在此优化的基础上得到了抑制剂的几何结构参数、电子结构参数以及静电势等信息.
2.2 分子力学优化
基于Filobasidiella neoformans IGPD酶的晶体结构(PDB库号:1RHY),在Amber力场下,通过赋氢原子坐标确定IGPD酶构象的力场参数.利用Insight II 2003程序包11分子力学优化软件Discover12对IGPD酶空间结构进行优化处理:选择Amber力场,依次利用最陡下降(收敛判据0.08 kJ·mol-1,收敛步长15000)、共轭梯度(收敛判据0.04 kJ·mol-1,收敛步长20000)优化方法,在考虑溶剂(水)效应的情况下,设定溶剂(水)的介电常数为80,采用GBMV(Generalized Born with Molecular Volume)模型进行分子力学优化,最终确定IGPD酶的理论空间构象.
2.3 分子对接及常温动力学优化
基于密度泛函方法优化获得的抑制剂a-g的稳定构象(图2),以及通过分子力学模拟获得的IGPD酶优化稳定构象,在考虑溶剂效应的情况下,通过对IGPD酶构象进行网格分析,借助Discovery studio 2.1中分子对接模块Ligand_fit13对抑制剂a-g与IGPD作用复合物结构进行分析,对接研究范围设定为0.80 nm.对接过程中,IGPD的格点盒子设定为6.30 nm×6.70 nm×7.40 nm,格点间距为0.05 nm,利用Lamarckian遗传算法,通过局域能量搜索与遗传算法相结合,以半经验势函数作为能量打分函数,对IGP及小分子抑制剂作用位置进行全局搜索,通过van der Waals作用能、静电作用能以及分子间氢键确认复合物的初始结构.
在Amber力场下,考虑溶剂(水)效应,首先对确定的复合物结构进行了分子力学优化(条件同上);进而进行常温分子动力学模拟分析:14设定模拟温度为300 K,溶剂采用TIP3P水模型,利用分子力学优化获得的模型进行20 ns的分子动力学模拟使体系达到平衡,收集20 ns的分子动力学模拟数据.在数据收集过程中,每隔20 ps记录一次模拟体系的构象.模拟中,采用Shake方法来约束体系中与氢原子有关的键伸缩,范德华相互作用的阈值(cut off)设定为1.00 nm,而静电相互作用采用Ewald加和法处理.在考察体系骨架的均方根偏差(RMSD)值的变化趋于稳定后,最终确定抑制剂与IGPD酶作用的结构特征.
3 结果与讨论
3.1 IGP及IGPD抑制剂分子的几何结构特征
化学分子的几何构型对分子中各原子的电荷分布、分子极性等均有较大的影响,进而将影响化合物的药效.15在没有化学分子构型的晶体结构数据的情况下,通常采用对化合物的低能态优势构象进行讨论.本研究中对IGP及IGPD抑制分子a-g进行优化,并经过振动频率分析无虚频,表明优化获得的上述分子构型确为可能的最低能优势构象.几何优化表明IGP及IGPD抑制分子a-g的分子结构具有类似的几何特征,即呈椅式,化合物IGP的构型示意图如图3所示.含有氮原子的芳香性五元环位于椅式构象的头部,烃链为椅背,而磷酸根位于椅式构象的底部.
3.2 IGPD构象优化及活性位点的构象特征
蛋白质中带电氨基酸的侧链在溶剂作用下容易发生电离,如Asp和Glu中的羧基、Lys和Arg中的氨基和胍基,His中的氨基.不同物种的IGPD蛋白多序列结构比对结果3表明,IGPD的C端包含(D/ N)XHHXXE模体,构成其与底物作用的核心区域.通过力学优化获得的IGPD构象如图4所示.进一步通过主链碳原子均方根位移对理论构象进行评判,其与晶体结构下的IGPD主链碳原子RMSD为0.029 nm,结果表明选择的分子力场以及相关的经验参数是合适的.

图3 利用B3LYP方法在6-31G**基组下获得的IGP构象示意图Fig.3 Conformation schematic diagram of IGPunder 6-31G**basis set using B3LYPdensity functional theory
借助Delphi程序对IGPD的C端模体(D/N)XHHXXE的表观静电分布情况进行了计算.图5给出了在考虑溶剂效应的前提下获得的IGPD的C端模体(D/N)XHHXXE的表观静电分布模式.可以看出, XHHX表观静电分布表现为正电;尽管谷氨酸(E)带有很强的负电,然而由于周围环境的影响,使得其负电性很弱,表观趋于中性,在靠近XHHX处拥有正电性.进而提示,对于抑制剂而言,磷酸根离子强的负电荷会与XHHX接触,而杂环部分与XE接触.
3.3 IGPD抑制剂与IGPD通过非键作用形成复合物的作用模式分析
基于IGPD优化的理论构象以及通过量化计算获得的IGPD抑制剂稳定构象,利用分子对接程序获得了IGPD与其抑制剂相互作用的复合物结构,并通过常温动力学模拟对获得的复合物结构进行了优化.图6(a)给出了抑制剂g与IGPD相互作用的复合物构象.
蛋白与抑制剂的作用主要是蛋白活性位点残基与抑制剂的相互作用.16抑制剂a、d由于其连接含氮杂环及PO(OH)2的脂肪链较b、c、e、f、g长,不能很好地识别C端模体,而是通过空间位阻效应阻断IGPD的活性,而b、c、e、f、g的脂肪链长度刚好嵌在T129R130(T、R分别代表蛋白IGPD的129、130位氨基酸)与E164
-H168之间,磷酸根离子主要识别IGPD的而含氮杂环与T129R130作用.T129R130位置的表观静电势为正,含氮杂环的原子电荷越负,二者的结合越好.

图4 基于IGPD晶体结构(PDB库号:1RHY),通过分子模拟、力学优化获得的三维理论IGPD空间构象飘带结构Fig.4 Three dimensional(3D)theoretical ribbonstructure of IGPD obtained by molecular simulation and mechanical optimization based on its crystal structure (PDB code:1RHY)

图5 用Delphi软件得到的IGPD蛋白C-端模体(D/N) XHHXXE的表观静电分布示意图Fig.5 Surface electrostatic distribution map of C-terminal motif(D/N)XHHXXE in IGPD protein using Delphi softwareThe red is the negative,the blue is the positive and the white was the neutral region.The green line denotes the main-chain carbon atom orientation of the IGPD,and the ball and stick modal denote the heavy atoms of the motif.
为了更准确地反映IGPD抑制剂与IGPD作用的情况,利用距离几何学、分子间氢键形成理论以及复合物相互作用的结合能对抑制剂a-g与IGPD相互作用模式进行了分析.定义距离IGPD抑制剂周围0.50 nm范围内的氨基酸为IGPD抑制剂识别IGPD的关键区域(图6(b)给出了抑制剂g识别IGPD的关键位置).表1给出了抑制剂a-g与IGPD相互作用过程中的结合能、分子间氢键形成数目、抑制剂识别IGPD的关键区域.
从图6及表1可以看出,IGPD抑制剂识别IGPD的活性口袋的氨基酸组成包含T129、R130、E164、N165、N166、H167、H168等,与晶体结构提示的活性区域一致.3从表1可以看出,抑制剂a、d由于其脂肪链长度一致而识别区域类似,而b、c、e、f、g的识别区域相似.

图6 IGPD蛋白与抑制剂g相互作用的复合物结构Fig.6 The 3D complex structure of IGPD protein and inhibitor g(a)The ribbon struture denotes the main-chain carbon atom orientation of the IGPD and the stick model denotes the heavy atoms of g, displaying with pymol software based on the optimized complex structure of IGPD and compound g;(b)The local model of the IGPD-g complex,where the pink line denotes the compound g,the green line denotes the main-chain carbon atom orientation of the IGPD and the yellow line denotes the identified domain in IGPD by compound g.The identified domain in IGPD was determined using distance geometry method and intermolecular hydrogen bond forming theory under InsightII 2003 software.
为了更清楚地分析抑制剂与IGPD的作用,图7给出了抑制剂a、g与IGPD作用的局域模式图.
从图7可以看出,抑制剂与IGPD相互作用以分子间氢键作用为主,van der Waals等疏水作用为辅.抑制剂a与IGPD作用中,抑制剂a的N4与IGPD中E171的羧酸根离子OE1形成距离为0.31 nm氢键,抑制剂a的磷酸根离子中的O18与IGPD中N165的侧链酰胺的氮原子N12形成距离为0.30 nm氢键;抑制剂a的脂肪链与IGPD中的H167存在弱的疏水作用;抑制剂g与IGPD相互作用的分子间氢键形成情况:抑制剂g的N2与IGPD的K132的氧原子O形成距离为0.29 nm的氢键;N165的酰胺氧原子OD1与抑制剂g的O20形成距离为0.31 nm的氢键;N166的酰氨基氮原子ND2与抑制剂g的O19形成距离为0.31 nm的氢键;N166的主链氨基氮原子N与抑制剂g的O19形成距离为0.33 nm的氢键;H167的主链氨基氮原子N与抑制剂g的O20形成距离为0.29 nm的氢键;抑制剂g的O20与H167的咪唑环氮原子ND1形成距离为0.29 nm的氢键;同时抑制剂g与IGPD中的V133、H168、L105存在疏水作用,从以上结果可以看出,氢键形成数目、疏水作用与抑制剂活性增长是一致的.

表1 IGPD抑制剂a-g与IGPD相互作用能和相互作用模式分析Table 1 The calculated interaction energies and interaction mode analysis between IGPD and its inhibitors a-g
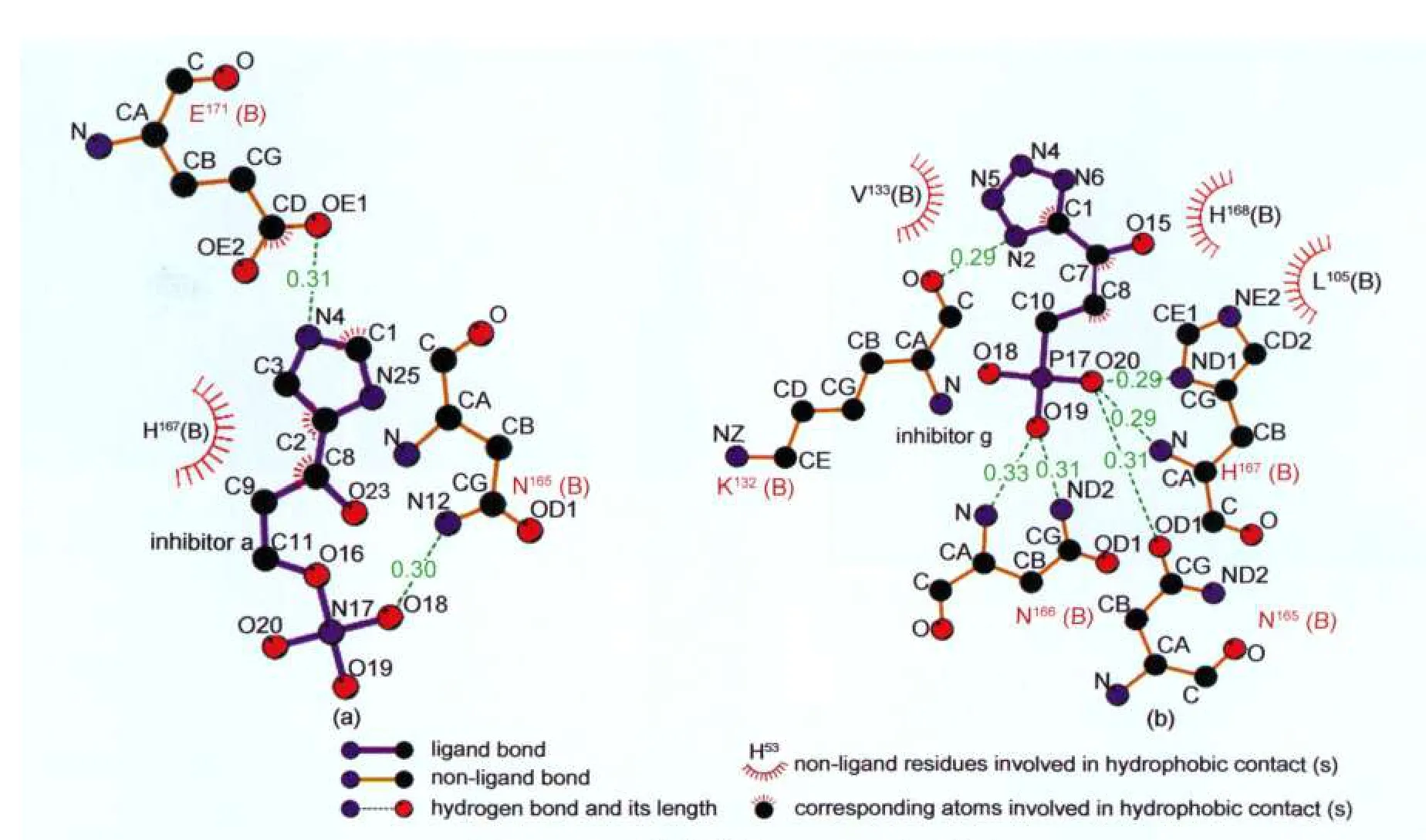
图7 IGPD与其抑制剂a(a)、g(b)作用模式Fig.7 Interaction information between IGPD and its inhibitor a(a)and g(b)Hydrogen bond lengths are in nm.
3.4 IGPD抑制剂与IGPD可能的亲电反应基础
按照量子化学前线分子轨道理论,化学小分子的前线分子轨道(最高占据分子轨道HOMO、最低非占据分子轨道LUMO)及其附近的分子轨道对化学分子的反应活性影响最大.17一般的,如果HOMO及其附近的占据分子轨道的能级较高,表明HOMO附近的占据轨道具有强的供电子能力,有利于亲核反应;18如果LUMO及其附近的空轨道的能级较低,表明LUMO附近的空轨道具有强的受电子能力,有利于亲电反应.19IGPD与其底物IGP作用机制(图1)的设想提示我们,当IGP或抑制剂与IGPD发生作用时,电子的流向是从IGPD的HOMO及其附近轨道流向IGP或抑制剂的LUMO及其附近的轨道.因此,对于IGP及其抑制剂应具有强的受电子能力,才能保证IGPD与其发生作用进而引发之后的反应.
依据密度泛函B3LYP方法选择6-31G**优化获得的抑制剂a-g稳定构象的前线分子轨道能量分布如表2所示.从表2可以看出,随着IGPD抑制剂结构的变化,其LUMO及其附近轨道的能级随着其IC50的降低而降低,而HOMO及其附近轨道的能量则随着IC50的降低而升高,表明电子的流向与设想一致,即抑制剂与IGPD发生作用时,电子从IGPD的HOMO及附近轨道流向抑制剂的LUMO及其附近轨道,而抑制剂的HOMO及其附近轨道的电子将稳定存在于抑制剂分子中,这可能是抑制剂对IGPD产生抑制活性的分子基础,同时也提示我们在筛选高活性的抑制剂过程中,其前线轨道的能级占据重要地位.20

表2 IGPD抑制剂的HOMO-1,HOMO,LUMO, LUMO+1轨道能量Table 2 The HOMO-1,HOMO,LUMO,LUMO+1 orbital energies of IGP(IGPDʹS substrate)inhibitors

表4 IGPD抑制剂含氮五元芳香环原子电荷布居分析Table 4 Atomic charge population analysis of nitrogen-containing five-ring in IGPD inhibitor

表3 IGPD-抑制剂(a-g)作用复合物中抑制剂的HOMO-1,HOMO,LUMO,LUMO+1轨道能量Table 3 The HOMO-1,HOMO,LUMO,LUMO+1 orbital energies of IGPD inhibitors from the complex of IGPD and its inhibitors(a-g)

图8 IGPD抑制剂a(a)、g(b)最低空轨道电子云密度空间分布情况Fig.8 Electronic clouds density distribution in the lowest unoccupied molecular orbital of the IGPDʹs inhibitor a(a)and g(b)
为了更清晰地判别IGPD抑制剂与IGPD可能的亲电反应,我们利用获得的稳定复合物(抑制剂与IGPD作用)结构,提取抑制剂构象,利用密度泛函B3LYP方法选择6-31G**基组对其进行单点计算,确定其前线轨道数据(表3),并对IGPD抑制剂a、g的LUMO轨道的空间分布进行了分析,见图8.
从表3可以看出,在抑制剂与IGPD作用后, LUMO轨道的能级相对于抑制剂的稳定构象下的LUMO轨道能级进一步降低,有利于电子的流入; HOMO轨道能级变化不大.以抑制剂IC50对应的浓度取常用对数为纵坐标,以表3列出的LUMO与HOMO能级比值(ELUMO/EHOMO)为横坐标,建立拟合关系,确定的方程为y=-0.5296x+4.0457(R2=0.9698).
从图8给出的抑制剂a、g的LUMO轨道分布可以看出,抑制剂的最低空轨道(LUMO)中电子云密度集中于含氮杂环,抑制剂g的电子云密度分布更为集中,更易于亲电反应.
为进一步探讨IGPD抑制剂分子中电荷分布状况对其活性的影响,利用Gaussian 03程序在密度泛函B3LYP方法下,选择6-31G**基组对IGPD抑制剂a-g的Mulliken电荷、自然键轨道电荷、21约束静电势方法(RESP)22下的原子电荷分布进行了研究.为了避免电荷分配中的任意性因素,综合考虑了Mulliken电荷布居、23自然键轨道(NBO)、24RESP下的电荷布居,22结果见表4.
从表4可以看出,随着IGPD抑制剂中的官能团——含氮五元芳香环中N原子的数目增多,含氮杂环的原子电荷布居(Mulliken、NBO、RESP)从正电荷向负电荷变化,IGPD抑制剂的活性提高.对于官能团PO(OH)2一侧,原子电荷分布类似,无论连接脂肪链,还是通过氧原子连接,均不影响整体电荷的分布.结果表明,官能团PO(OH)2在与酶作用中,识别位置是固定的,而另外一侧的含氮五元环由于电荷分布的差异而识别的位置稍有变化并进而影响其功能.
4 结论
借助IGPD的晶体结构(PDB库号:1RHY),通过牛顿力学优化、分子对接以及量子化学密度泛函理论研究了IGPD与其抑制剂之间的相互作用.依据计算结果以及含氮五元环芳香族抑制剂的活性(IC50)分析可以看出,抑制剂的LUMO及其附近轨道能级、活性官能团原子电荷分布(Mulliken电荷、自然键轨道电荷、考虑RESP的电荷分布)、IGPD活性位点的静电性质、抑制剂与IGPD的结合方式对于IGPD与其抑制剂的相互作用发挥重要的作用.基于此,对于数据库的高通量筛选结果进行进一步的评判及合成具有重要的指导意义,并将有利于发现并设计新型的靶向IGPD的抑制剂.
(1) Xiao,Y.J.;Wang,J.G.;He,F.Q.;Li,Z.M.Pesticides 2005,44 (10),433.[肖勇军,王建国,何凤琦,李正名.农药,2005,44 (10),433.]
(2)Tada,S.;Hatano,M.;Nakayama,Y.;Volrath,S.;Guyer,D.; Ward,E.;Ohta,D.Plant Physiol.1995,109,153.
(3) Sinha,S.C.;Chaudhuri,B.N.;Burgner,J.W.;Yakovleva,G.; Davisson,V.J.;Smith,J.L.J.Biol.Chem.2004,279,15491.
(4) Mori,I.;Fonne-Pfister,R.;Matsunaga.S.;Tada,S.;Kimura,Y.; Iwasaki,G.;Mano,J.;Hatano,M.;Nakano,T.;Koizumi,S.; Scheidegger,A.;Hayakawa,K.;Ohta,D.Plant Physiol.1995, 107,719.
(5)Mori,I.;Iwasaki,G.;Kimura,Y.;Matsunaga,S.;Ogawa,A.; Nakano,T.;Buser,H.P.;Hatano,M.;Tada,S.;Hayakawa,K. J.Am.Chem.Soc.1995,117,4411.
(6) Mori,I.,Iwasaki,G.;Hayakawa,K.Yuki Gosei Kagaku Kyokaishi 1996,54,514.
(7) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.01;Gaussian Inc.:Pittsburgh,PA,2004.
(8) Becke,A.D.J.Chem.Phys.1993,98,5648.
(9) Frisch,M.J.;Pople,J.A.;Binkley,J.S.J.Chem.Phys.1984, 80,3265.
(10) Wang,Y.L.;Wu,G.S.Acta Phys.-Chim.Sin.2008,24,552. [王溢磊,吴国是.物理化学学报,2008,24,552.]
(11) Insight II,Version 98.0;Molecular Simulation Inc.:San Diego, CA,1998.
(12) Discover 3 User Guide,Version 98.0;Molecular Simulation Inc.:San Diego,CA,1998.
(13) Discovery Studio,version 2.0;Molecular Simulation Inc.:San Diego,CA,2007.
(14)Xu,Y.C.;Shen,J.H.;Luo,X.M.;Shen,X.;Chen,K.X.;Jiang, H.L.Science in China Series B-Chemistry 2004,34,177. [许叶春,沈建华,罗小民,沈 旭,陈凯先,蒋华良.中国科学B辑:化学,2004,34,177.]
(15)Qu,X.B.;Su,Z.M.;Hu,D.H.;Bao,Y.L.;Meng,X.Y.;Wu, Y.;Li,Y.X.Chem.J.Chin.Univ.2009,30,1402.[曲晓波,苏忠民,胡冬华,鲍永利,孟祥颖,乌 垠,李玉新.高等学校化学学报,2009,30,1402.]
(16)Liu,C.L.;Li,C.H.;Chen,W.Z.;Wang,C.X.Acta Phys.-Chim.Sin.2005,21,1229.[刘春莉,李春华,陈慰祖,王存新.物理化学学报,2005,21,1229.]
(17)Chen,P.Q.;Liu,X.H.;Sun,H.W.;Wang,B.L.;Li,Z.M.;Lai, C.M.Acta Chim.Sin.2007,65,1693.[陈沛全,刘幸海,孙宏伟,王宝雷,李正名,赖城明.化学学报,2007,65,1693.]
(18) Jiang,Y.;Guo,Z.R.Chin.J.Org.Chem.2004,24,1640. [蒋 毅,郭宗儒.有机化学,2004,24,1640.]
(19) Wu,H.;Huang,Z.Z.;Chen,X.J.;Huang,Z.P.;Zheng,Y. Chinese Journal of Biochemistry and Molecular Biology 2007, 23,959.[吴 洪,黄真珠,陈秀娟,黄增平,郑 勇.中国生物化学与分子生物学报,2007,23,959.]
(20) Cui,B.Q.;Zhao,D.X.;Yang,Z.Z.Acta Chim.Sin.2007,65, 2687.[崔宝秋,赵东霞,杨忠志.化学学报,2007,65,2687.]
(21) Chen,W.K.;Zhang,Y.F.;Ding,K.N.;Xu,Y.J.;Li,Y.;Li,J. Q.Chin.J.Struct.Chem.2004,23,337.[陈文凯,章永凡,丁开宁,徐艺军,李 奕,李俊篯.结构化学,2004,23,337.]
(22)Zhao,X.;Wang,S.;Xu,X.H.;Huang,X.R.Acta Chim.Sin. 2009,67,1835.[赵 熹,王 嵩,徐晓华,黄旭日.化学学报, 2009,67,1835.]
(23) Liu,J.C.;Zhang,X.M.;Chen,M.A.;Tang,J.G.;Liu,S.D. Acta Phys.-Chim.Sin.2010,26,163.[刘建才,张新明,陈明安,唐建国,刘胜胆.物理化学学报,2010,26,163.]
(24) Pan,G.X.;Ni,Z.M.;Li,X.N.Acta Phys.-Chim.Sin.2007,23, 1195.[潘国祥,倪哲明,李小年.物理化学学报,2007,23, 1195.]
January 26,2011;Revised:May 5,2011;Published on Web:June 13,2011.
Molecular Simulation of the Interaction between Imidazole Glycerol Phosphate Dehydrase and Nitrogen-Containing Heterocyclic Phosphate Inhibitors
SHEN Tao1DU Feng-Pei1LIU Ting1YAO Guang-Wei1WU Zheng1FANG Meng-Meng1XU Xiao-Jie2LU Hui-Zhe1,*
(1Institute of Science and Technology,China Agricultural University,Beijing 100193,P.R.China;2Institute of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China)
A series of nitrogen-containing heterocyclic compounds as imidazole glycerol phosphate dehydrase(IGPD)inhibitors were successfully screened based on IGPD substrates;however,the mechanism is not clear.In this study,the B3LYP density functional theory method with the 6-31G**basis set as implemented in the Gaussian 03 program was selected to optimize the nitrogen-containing heterocyclic phosphates.These complex structures were constructed using molecular docking and optimization.The mode of interaction was discussed with regards to their electronic structures(frontier orbital energies and composition,the atomic charges,the natural bond orbital,etc.)and complex spatial structures(recognition functional domains of the inhibitor of IGPD,intermolecular hydrogen bonding,van der Waals interactions,etc.).The charge distribution of the nitrogen-containing heterocycle,the phosphate ion charge distribution,and the frontier orbital LUMO energy levels of the inhibitor were determined to be intrinsic factors that affect inhibitor activity.The conclusion of our study will provide valuable information for the screening and optimization of new herbicides targeted at IGPD.
Imidazole glycerol phosphate dehydratase;Inhibitor;Molecular docking;Mechanical optimization; Density functional theory;Frontier orbital
O641
*Corresponding author.Email:luhz2008@yahoo.com.cn;Tel:+86-10-62732507.
The project was supported by the National Natural Science Foundation of China(20972184)and Chinese Universities Scientific Fund(2009JS38, 2011JS036).
国家自然科学基金(20972184)和中央高校基本科研业务费专项资金(2009JS38,2011JS036)资助项目
猜你喜欢
世界农药(2019年4期)2019-12-30
西南石油大学学报(自然科学版)(2018年2期)2018-06-26
现代食品(2016年24期)2016-04-28
中国粮油学报(2016年1期)2016-02-06
中国资源综合利用(2016年7期)2016-02-03
中国资源综合利用(2016年12期)2016-01-22
河北大学学报(自然科学版)(2015年1期)2015-02-27
应用化工(2014年1期)2014-08-16
影像科学与光化学(2014年3期)2014-03-11
中国粮油学报(2014年8期)2014-02-06
