
磷在P-ZSM-5沸石中存在的形态
2011-11-30 10:50孙迎新赵立峰
物理化学学报 2011年8期
杨 静 孙迎新 赵立峰 孙 淮
(上海交通大学化学化工学院,上海200240)
磷在P-ZSM-5沸石中存在的形态
杨 静 孙迎新 赵立峰 孙 淮*
(上海交通大学化学化工学院,上海200240)
用密度泛函理论和ONIOM(our own N-layer integrated molecular orbital molecular mechanics)方法研究磷改性的ZSM-5沸石中含磷基团的可能存在形态.计算的反应焓和自由能数据表明P-ZSM-5沸石中以磷进入骨架和在骨架外的形成磷酸根离子对是合理的稳定结构.而且,计算结果表明离子对模型F和G更适合在室温下存在,磷进入骨架的酸性结构Cʹ在高温下更稳定,而磷进入骨架的结构C对温度变化不敏感.计算得到的27Al,31P,29Si化学位移、酸性的变化趋势和结构参数与相关实验数据吻合.
H-ZSM-5沸石;磷改性;ONIOM方法;密度泛函理论
1 引言
ZSM-5分子筛是石油化工领域广泛使用的催化材料之一.因为催化剂一般都在高温下使用,如何提高ZSM-5沸石的水热稳定性是主要研究课题之一.1,2在高温下,经过若干反应循环后沸石中的铝原子被排斥到骨架之外,最终使催化剂失去活性.磷改性能提高ZSM-5分子筛的水热稳定性而延缓脱铝过程的发生.3,4不同Si/Al比的ZSM-5分子筛可以用各种含磷化合物进行处理,例如H3PO4,5,6NH4H2PO4,6P(C6H5)37和P(OCH3)38等等,生成磷改性的ZSM-5沸石(P-ZSM-5沸石).P-ZSM-5沸石通常用来催化甲醇转化制轻烯烃,9,10芳烃的烷基化或者歧化,11,12并且它们也是在流化催化裂化(FCC)中促进氢烯烃形成的重要添加剂.13
许多的实验工作例如X射线衍射(XRD),NH3-程序升温脱附(TPD),吡啶-红外(IR)光谱,29Si、27Al、31P核磁共振谱(NMR)都被用来研究P-ZSM-5沸石的结构和性质.因为磷的加入能减少Brönsted酸性位点,Caro等14认为骨架脱铝而形成磷铝酸盐.Seo和Ryoo15把酸性位点的减少归咎于形成了和磷相互作用的八面体铝.Lischke等16,17报导在水热处理后P-ZSM-5沸石的Brönsted酸性位点得以保存,并且在用热水洗涤后恢复了额外的酸性位点.Zhuang等18提出磷取代骨架硅形成(SiO)xAl(PO)4−x物种而导致了脱铝.Blasco等6提出离子对模型,在这个模型中骨架铝被骨架外的磷酸阳离子所保护.de Menezes等5报导了许多类型的铝物种.除了骨架四配位铝,还有大量的骨架外扭曲的四配位铝和六配位铝. Zhao等19表明磷改性不仅提高了水热稳定性,而且增加了抗结焦性能,有助于提高C4烯烃裂解的催化性能.他们也提出扭曲的四配位铝或者五配位铝仍可成为活性位点.近来,Xue等20的研究表明在1073 K时P-ZSM-5沸石可能由于磷与骨架外铝相互作用形成新的稳定的酸性位点.总之,关于磷如何与沸石相互作用,磷是否占据骨架,磷在骨架里的位置等仍然存在着争议.
理论计算方法可以从微观尺度上对比研究不同结构的相对稳定性.已有一些用量子力学密度泛函理论(DFT)研究不同元素改性ZSM-5沸石的工作报道.Broclawik等21计算了Ga改性的ZSM-5沸石. Zhou等22,23研究了Mo和La改性ZSM-5沸石.然而,文献中尚未看到对磷改性ZSM-5沸石进行系统的理论计算研究.本文采用密度泛函理论和ONIOM方法24研究磷改性的P-ZSM-5沸石中含磷基团.研究的主要目的是理解磷是怎样和沸石相互作用和评估各种可能的存在形态,并计算了可能结构的热力学稳定性及它们的相对酸性和水热稳定性,通过这些计算希望揭示P-ZSM-5沸石水热稳定性和催化性能增加的原因.
2 计算方法和模型
用H-ZSM-5沸石中的含Brönsted酸性中心的簇模型作为基本模型.用8T簇模型(模型A,见图1)表示全硅沸石.沸石晶体结构的T12位点被选做Al取代的位置.25,26通过用铝取代其中的一个硅原子,再在桥氧上添加一个氢原子,获得了酸性模型(模型B,见图1).8T模型可以用来进行全从头算优化.为了消除计算尺寸的影响,我们用了更大的含酸性中心的模型(46T和128T,见图2).除了模型A和B,在这个基础上构建磷改性后的模型,细节将在后面描述.
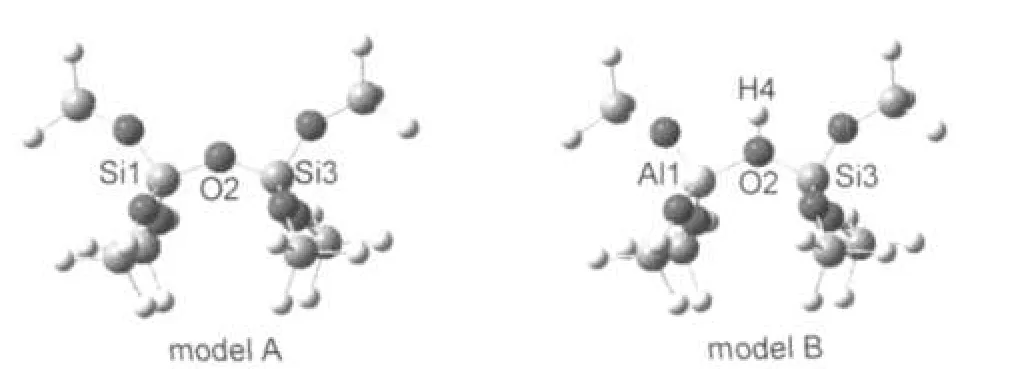
图1 8T全硅(模型A)和铝硅沸石(模型B)Fig.1 8T cluster models for silica(modelA)andaluminosilicate(model B)zeolites

图2 46T和128T铝硅沸石(延展的模型B)Fig.2 46T and 128T models for aluminosilicate zeolite (extended model B)
用DFT方法优化了模型化合物在沸石环境限制下的结构.对于8T模型,结构优化都是使用B3LYP(Beeke-3-Lee-Yang-Parr)泛函和6-31G(d)基组.初始结构从ZSM-5沸石的晶体结构27中截取,尾端未饱和的硅沿着Si―O键的方向用氢原子进行饱和,Si―H键的键长固定在0.15 nm.沸石环境的限制用固定端部的―SiH3基团的位置实现,其它原子的位置在结构优化的过程中都是自由变化的.对于46T和128T模型,我们用ONIOM方法28进行了计算.在这些计算中,内层(8T区域)是在B3LYP/ 6-31G(d)水平上进行,而外层则用UFF(universal force field)力场29处理.所有外层的原子根据原晶体的结构确定.这种计算方法在前人的工作30中都使用过,并且得出的结果能够很好地解释实验结果.
在优化结构的基础上进行正则分析以确定能量最小点.在给定温度下计算了模型化合物的热力学性质(包括生成焓和自由能),计算中振动频率的校正因子是0.9804.31计算了同性屏蔽常数(σ),进而得到化学位移(δ).对于27Al和31P化学位移,分别采用Al(NO3)3和H3PO4做标样计算:
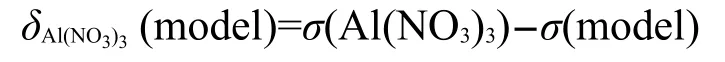

图3 可能的磷改性ZSM-5沸石模型C-IFig.3 Proposed models C-I for P-ZSM-5 zeolites

对于29Si化学位移,我们用Moravetski等32提出的方案进行计算,在这个方案中Si(OH)4被用作参考样来计算模型化合物的化学位移:
δTMS(model)=σ(Si(OH)4)-σ(model)+δTMS(Si(OH)4)其中δTMS(Si(OH)4)为实验化学位移值(-72.0).33
溶剂化自由能用极化连续介质模型模(PCM)及默认的参数计算.34-36
大部分计算用Gaussian 03软件37执行,关于同性屏蔽常数的计算用Turbomole软件38完成,以加快计算效率.
3 结果与讨论
3.1 磷可能的存在形态
在模型B的基础上构建了模型C到I,见图3.在模型C中磷取代了骨架中的硅,这个模型由Zhuang等18提出来的.因为这个模型没有酸性中心,我们称之为骨架内的磷物种.模型Cʹ是模型C的一个变化,在这个模型里磷基团提供了一个酸性位点.这个模型中有一个双键氧原子,它能与骨架外或骨架内的铝相配位.模型D是通过加一个磷酸分子后,脱去一个水分子形成的,这个模型最初由Védrine等9, Kaeding和Butter3提出.模型E对应于同一个机理,但是脱去了两个水分子,这个模型是由Rahman等7提出来的.在模型D和E中磷代替了桥氢和桥氧直接相连.模型F和G是离子对模型,在这个模型里阳离子或和桥氧负离子相互作用.8,39

图4 在B3LYP/6-31G(d)方法下优化得到的8T磷改性ZSM-5沸石模型C-GFig.4 B3LYP/6-31G(d)optimized structures of the 8T models C-G representing phosphorus impregnation ZSM-5 zeolites
用上述计算方案,我们获得了优化的8T模型C到G.这些优化的模型显示在图4中.然而,计算无法得到模型H和I的优化结构.这个模型先前也有几个工作提出.40,41尽管这两个模型从化学结构上看是合理的,但沸石骨架的几何结构阻止了H2PO3基团的插入.因为沸石中铝原子和硅原子之间的距离大约是0.30-0.35 nm,而要插入一个H2PO3基团至少需要0.40-0.50 nm.在实验文献中,还讨论到磷物种有可能与非骨架铝物种相关,这涉及到的结构比较复杂,因为非骨架铝物种可能与水形成各种配位体,在本文中我们就不进行讨论.
由于上述模型分子具有不同的原子种类与个数,为了评估模型C到G的相对稳定性,根据前人的方案,42我们设计了以下6个假想的由模型B可以得到模型C-G分子的化学反应,并计算了这些反应的自由能和生成焓变.
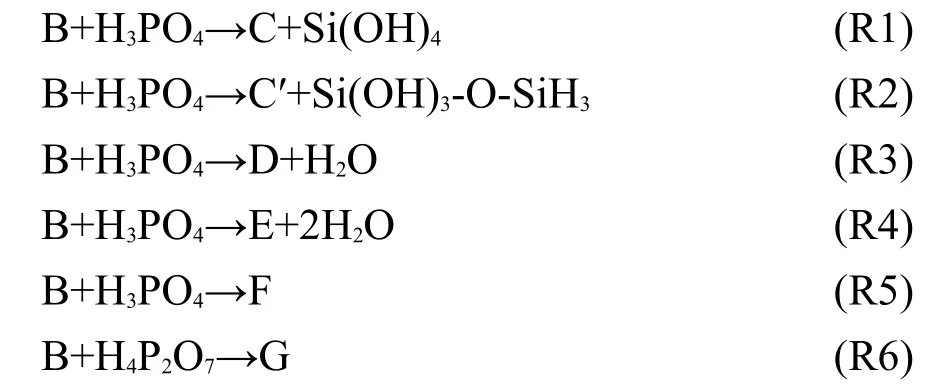
从298到1075 K用8T簇模型计算的生成焓ΔH和自由能变ΔG列于表1中.反应(R1)小量吸热,但是自由能变是负值,表明化合物C从热力学的观点来说是稳定的.反应(R2)是吸热反应,在室温下,自由能变是正值.然而,因为反应是熵增的,随着温度的增加,自由能变逐渐减少.在高温1075 K时,自由能变为负值(-50.0 kJ·mol-1).反应(R3)和(R4)大量吸热,在热力学上不利进行.反应(R5)和(R6)在整个温度范围都是大量放热;然而,因为反应是熵减的,自由能变ΔG依赖于温度.在高温下,反应(R5)和(R6)的自由能变成正值.

表1 在不同温度下计算的8T模型B生成模型C-G的热力学能量数据(单位:kJ·mol-1)aTable 1 Thermodynamic energy data(in kJ·mol-1)for the formation of models C to G from model B at different temperatures using 8T modelsa
模型D(R3产物)的不稳定是因为含磷的基团和骨架之间的空间排斥作用.仔细分析键长显示P5―O2键的键长(图4)是0.18 nm,这比磷酸中的一般值(大约0.16 nm)长了许多.模型E(R4的产物)不稳定是因为磷的共价结构.在这个模型中磷有三个键,其中两个是双键.一般来说,对于磷来说,锥形结构比四面体结构更不稳定.反应(R5)和(R6)释放大量热并不意外.阳离子H4PO4+和H5P2O7+的中心磷原子保持四面体共价结构,比较稳定,并且P―O键的键长均在通常范围内(0.158-0.159 nm).两个带相反电荷的基团之间的相互作用使复合物F和G(R5和R6的产物)非常稳定.
沸石环境对计算的热力学数据有明显影响,如表2中所示.因为模型Cʹ涉及到(或者骨架内或者骨架外)铝,反应(R2)没有包括在ONIOM计算中.对于其它的反应,计算的热力学能量的变化趋势类似于用8T簇模型的结果.对于反应(R3)到(R6),用46T和128T模型计算得到的生成焓和自由能变通常都低于用8T簇模型计算的结果.然而对于反应(R1),尺寸的大小没有起重大的作用.因为反应(R3)到(R6)的产物涉及骨架外的磷基团(图3,4中的模型D、E、F和G),但是反应(R1)的产物(模型C)是骨架内磷.沸石环境对于稳定骨架外磷基团的作用大于稳定骨架内磷基团的作用.

表2 对比298 K下用不同尺寸的模型B计算生成模型C-G的热力学能量数据(单位:kJ·mol-1)Table 2 Comparison of the calculated thermodynamic energy data(in kJ·mol-1)for the formation of models C to G from model B with different model sizes at 298 K
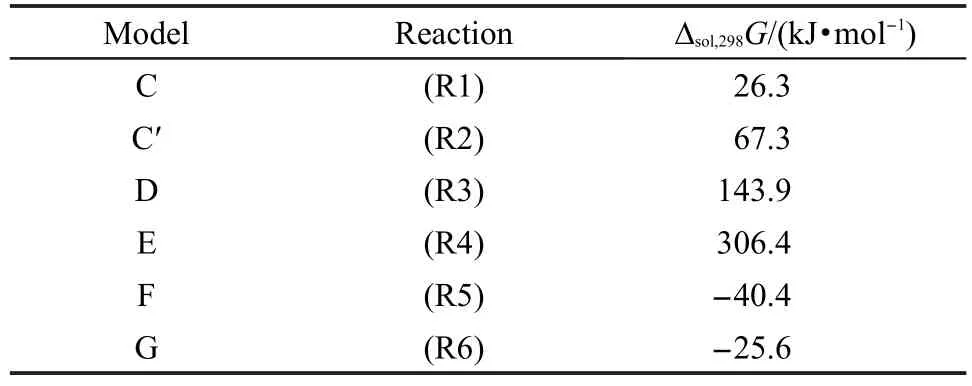
表3 由PCM模型计算水中的8T模型B生成模型C-G的溶剂化自由能变Table 3 Free energy differences for the formation of models C to G from 8T model B using PCM model in water solvent
文献34-36用PCM模型计算了在水中的反应(R1)-(R6)的自由能变,可以用来考察溶剂的影响.因为方法的限制,这些计算我们只用8T模型进行,结果列于表3中.可以看到溶剂效应显著,反应(R1)的自由能增加到26.3 kJ·mol-1,反应(R3)和(R4)的自由能分别增加到143.9和306.4 kJ·mol-1,反应(R5)和(R6)的自由能绝对值分别减少到40.4和25.6 kJ· mol-1.这表明反应(R5)和(R6)在溶剂中更容易进行.所有涉及的化合物的溶剂化自由能列于表4中.对于大多数的含硅模型化合物,溶剂化自由能是正值(>100 kJ·mol-1),表明他们是不溶于水的;两个离子对化合物在在溶液中更稳定(对于模型F是39.6 kJ· mol-1,对于模型G是-14.1 kJ·mol-1).对于焦磷酸H4P2O7和硅酸H4SiO4,溶剂化自由能是负值,表明这些化合物在水中的溶剂化是自发进行的.
3.2 化学位移
计算的化学位移列于表5中.对于模型A、B、F和G,我们计算了和桥氧相连的硅原子的29Si化学位移(图4中的Si3).模型A的29Si化学位移是-115.8,这与实验值-1136或-114.818比较接近,通常在固态NMR谱中它被归属于(Si(4Si))结构单元.对于模型B,计算的29Si位移是-110.9,它在实验值-1056或-107.418的附近并且归属于Si(3Si,1Al)结构单元.对于模型F和G,计算的29Si化学位移分别是-108.4和-109.0.从所有计算的29Si化学位移,我们可以看出用6-31G(d)基组计算的值一般都低于实验值,而用def-TZVP基组优化得到的值和实验值更接近.32

表4 用PCM模型计算的水中的溶剂化自由能Table 4 Calculated solvation free energies using PCM model with water
对于模型B、C、F和G我们计算了27Al的化学位移.对于模型B,计算的位移值58.1和实验上观察到的54.018相近,这归属于四面体骨架铝.模型C的计算位移值是59.0.对于离子模型F和G,计算得到的化学位移63.4和62.4,在实验值(63.7)18的附近.
对于模型C、Cʹ、F和G计算了31P化学位移.对于模型C和Cʹ,计算值分别是-36.6和-43.5,都处于实验测得的宽谱带区域-30--40之间,在文献中这个谱带的解释是有争议的.Blasco等6报道的NMR谱在这个区域中有两个主要的峰-32和-40,他们把-32的峰归属于无定形磷铝酸盐,而-40的峰归属于多磷酸盐或者聚磷物种.然而,Zhuang等18推断在-30--40之间的宽信号归属于骨架中的各种(SiO)xAl(OP)4-x物种.为了探明这个宽信号的归属,我们通过构建两个延展的含磷模型进一步计算了“铝对(aluminum pair)”结构43,44(见图5)的化学位移.模型C1只有一个铝原子,而模型C2有两个铝原子.计算的31P化学位移对于模型C1是-41.4,对于模型C2是-30.7,这两个值都在这个有争议的区域中,计算结果支持磷进入骨架内的假定.
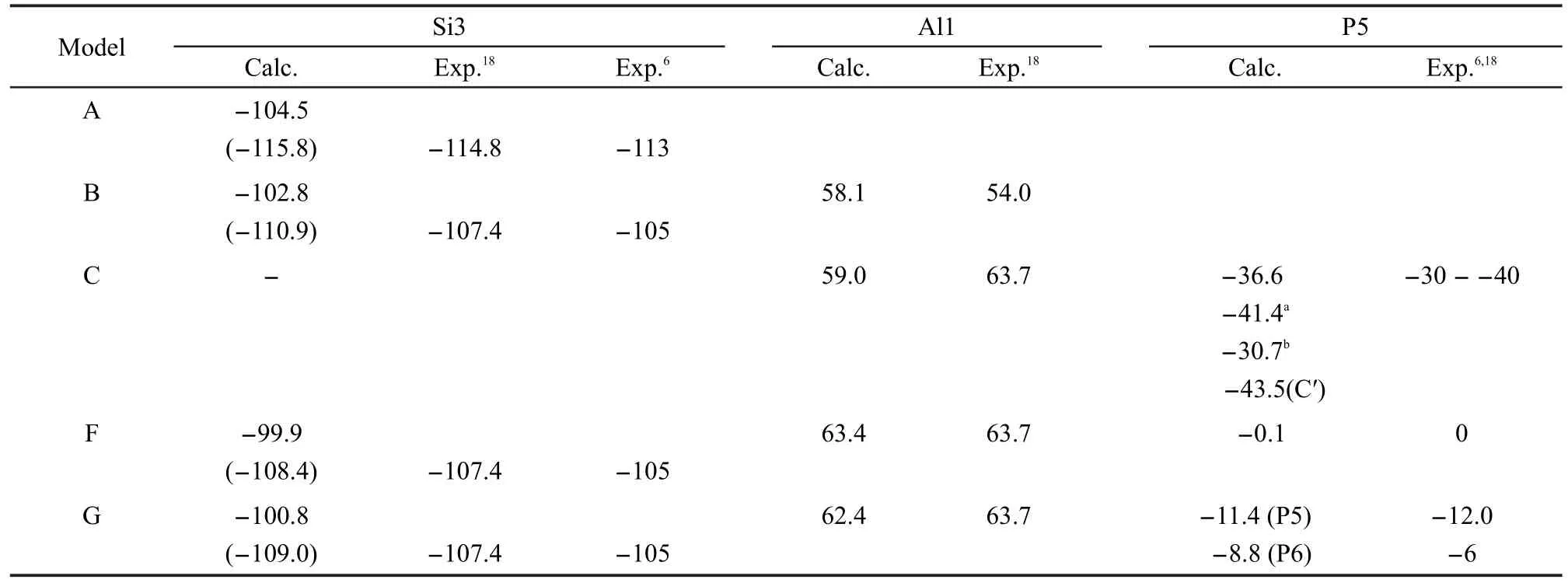
表5 计算的模型化合物的化学位移Table 5 Chemical shifts calculated for the model compounds
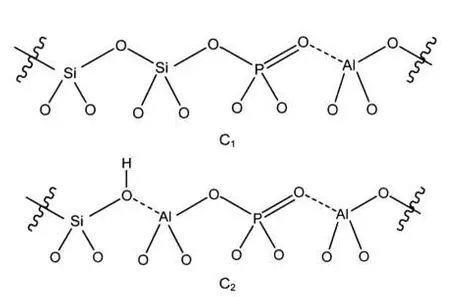
图5 有一个(C1)或者两个(C2)铝原子和一个磷原子在骨架中的P-ZSM-5沸石Fig.5 Extended models of P-ZSM-5 zeolite with one (C1)or two(C2)aluminum atoms and one phosphorus atom in the framework
对于模型F计算的31P化学位移是-0.1,对于单独的磷酸根阳离子我们获得了相似的值(-1.9).对于模型G,获得了两个位移(-8.8和-11.4),它们分别对应于焦磷酸盐末端和中部的基团,这和实验值(-6和-12)6,18相近.
3.3 酸性的变化
我们通过计算在模型B、Cʹ、F和G中羟基的脱质子化能,自然键轨道(NBO)电荷和伸缩振动频率考察了酸性的变化.在我们的计算中脱质子化能是指模型的能量减去脱质子的模型的能量,即ΔEDP=在模型B和Cʹ中只有一个羟基,模型F和G中有多个羟基,数据采用了平均值.计算的结果列于表6中.模型B的脱质子化能是1190.4 kJ· mol-1,模型Cʹ是1153.5 kJ·mol-1,模型F是1212.8 kJ·mol-1,模型G是1244.6 kJ·mol-1.计算的极化氢原子上的NBO电荷和脱质子化能紧密相关.在模型B中计算的O―H基团的伸缩振动频率是3519 cm-1,这和实验中得到的3600-3610 cm-1区域的谱带6相近.模型Cʹ的振动频率是3408 cm-1.模型F和G中羟基的振动频率分别是3602和3614 cm-1,这和实验报导的磷羟基的振动频率(3676 cm-1)6一致.模型F和G的酸性比模型B的酸性小,和实验结果一致.6

表6 8T模型B,Cʹ,F和G的脱质子化能(ΔEDP),NBO电荷(q)和羟基的伸缩振动频率(ν)Table 6 Deprotonation energies(ΔEDP),NBO charges(q), and stretch mode frequencies(ν)calculated for the hydroxyl groups in 8T models B,Cʹ,F,and G
吡啶通常用来探测沸石的酸性,它的振动频率出现在1400-1700 cm-1之间.我们优化得到了吡啶和模型化合物B、Cʹ、F和G相互作用的分子簇模型,用这些优化的结构我们计算了吡啶和模型化合物之间的结合能,吡啶的ν8a和ν19a振动模式的频率.45这些计算结果列于表7中.吡啶和模型B之间的结合能是-129.6 kJ·mol-1,这个值比吡啶和模型Cʹ之间的结合能(-204.1 kJ·mol-1)弱,但是强于吡啶与模型F和G之间的结合能(分别是-108.6和-77.3 kJ· mol-1).计算的频率和实验值46,47相近.吡啶的ν8a/8b和ν19a/19b振动频率按照下列顺序排列:Cʹ>B>F>G.

表7 吡啶和模型化合物的结合能(ΔEa)和对比实验和计算的吡啶特征振动频率Table 7 Binding energies(ΔEa)between pyridine and the model compounds and comparison of the experimental and calculated characteristic vibrational frequencies of pyridine
这些结果指示酸性的强度遵循下列的顺序: P-OH-Al(模型Cʹ)>Si-OH-Al(模型B)>P-OH(模型F和G).应该指出的是酸性的骨架内磷结构(模型Cʹ)提供最强的酸性中心.这和实验结果吻合,在Xue等20的文章中,在高温焙烧的条件下,酸性会有所增加.模型的酸性增加可以增加沸石的催化性能.
4 结论
计算的31P、29Si和27Al化学位移和实验报导的结果相一致.尽管许多重要的信号隐藏在宽信号中使得难以鉴定,但是计算为判断结构的合理性提供了依据.尤其是,我们发现31P信号在大约-32和-40处的分裂可能是因为磷进入骨架中分别有一个或者两个邻近的铝原子造成的.
通过计算脱质子化能、NBO电荷和羟基的振动频率发现,酸性变化与已知的实验数据一致.在磷改性之后,沸石的酸性依据形成不同的局域结构而变化.对于模型C没有强酸位点.模型Cʹ提供了一个强的酸性中心和催化性能.对于模型F或者G,酸性由磷羟基提供.在常温下,离子对模型F和G是最稳定的,这些结构有效地保护了酸性中心,而且材料的酸性很容易通过水洗恢复.
(1) Liu,Z.;Ouri,Y.;Qi,Y.;Zhang,S.;Zhang,Y.;Liu,Z. Microporous Mesoporous Mat.2006,93,205.
(2) Lourenço,J.P.;Fernandes,A.;Henriques,C.;Ribeiro,M.F. Microporous Mesoporous Mat.2006,94,56.
(3) Kaeding,W.W.;Butter,S.A.J.Catal.1980,61,155.
(4) Kaeding,W.W.;Chu,C.;Young,L.B.;Ourinstein,B.;Butter, S.A.J.Catal.1981,67,159.
(5)de Menezes,S.M.C.;Lam,Y.L.;Damodaran,K.;Pruski,M. Microporous Mesoporous Mat.2006,95,286.
(6) Blasco,T.;Corma,A.;Martínez-Triguero,J.J.Catal.2006, 237,267.
(7)Rahman,A.;Lemay,G.;Adnot,A.;Kaliaguine,S.J.Catal. 1988,112,453.
(8)Tynjälä,P.;Pakkanen,T.T.Microporous Mesoporous Mat. 1998,20,363.
(9) Védrine,J.C.;Auroux,A.;Dejaifve,P.;Ducarme,V.;Hoser,H.; Zhou,S.J.Catal.1982,73,147.
(10) Suzuki,K.;Kiyozumi,Y.;Matsuzaki,K.;Ikai,S.;Shin,S.Appl. Catal.1988,39,315.
(11) Dumitriu,E.;Bilba,N.;Hulea,V.;Oprea,S.Rev.Roum.Chim. 1989,34,983.
(12) Romannikov,V.N.;Tissler,A.J.;Thome,R.React.Kinet. Catal.Lett.1993,51,125.
(13) Degnan,T.F.;Chitnis,G.K.;Schipper,P.H.Microporous Mesoporous Mat.2000,35,245.
(14) Caro,J.;Bülow,M.;Derewinski,M.;Haber,J.;Hunger,M.; Kärger,J.;Pfeifer,H.;Storek,W.;Zibrowius,B.J.Catal.1990, 124,367.
(15) Seo,G.;Ryoo,R.J.Catal.1990,124,224.
(16) Lischke,G.;Eckelt,R.;Gjerschkewitz,H.;Parlitz,B.;Schreier, E.;Storke,W.;Zibrowius,B.;Oehlmann,G.J.Catal.1991, 132,229.
(17) Oehlmann,G.;Jerschkewitz,H.G.;Lischke,G.;Eckelt,R.; Parlitz,B.;Schreier,E.;Zibrowius,B.;Loeffler,E.Stud.Surf. Sci.Catal.1991,65,1.
(18) Zhuang,J.;Ma,D.;Yang,G.;Yan,Z.;Liu,X.;Liu,X.;Han,X.; Bao,X.;Xie,P.;Liu,Z.J.Catal.2004,228,234.
(19) Zhao,G.;Teng,J.;Xie,Z.;Jin,W.;Yang,W.;Chen,Q.;Tang,Y. J.Catal.2007,248,29.
(20)Xue,N.;Chen,X.;Nie,L.;Guo,X.;Ding,W.;Chen,Y.;Gu, M.;Xie,Z.J.Catal.2007,248,20.
(21)Broclawik,E;Himei,H.;Yamadaya,M.;Kubo,M.;Miyamoto, A.J.Chem.Phys.1995,103,2102.
(22) Zhou,D.;Ma,D.;Liu,X.;Bao,X.J.Chem.Phys.2001,114, 9125.
(23)Yang,G.;Wang,Y.;Zhou,D.;Zhuang,J.;Liu,X.;Han,X.; Bao,X.J.Chem.Phys.2003,119,9765.
(24)Maseras,F.;Morokuma,K.J.Comput.Chem.1995,16,1170.
(25) Schröder,K.P.;Sauer,J.;Leslie,M.;Catlow,C.R.A.Zeolites 1992,12,20.
(26) Stave,M.S.;Nicholas,J.B.J.Phys.Chem.1995,99,15046.
(27) van Koningsveld,H.;Jansen,J.D.;van Bekkum,H.J.Zeolites 1990,10,235.
(28) (a)Namuangruk,S.;Pantu,P.;Limtrakul,J.J.Catal.2004,225, 523. (b)Namuangruka,S.;Tantanak,D.;Limtrakul,J.J.Mol.Catal. A 2006,256,113.
(29) Rappe,A.K.;Casewit,C.J.;Colourll,K.S.;Goddard,W.A.; Skiff,W.M.J.Am.Chem.Soc.1992,114,10024.
(30) Lomratsiri,J.;Probst,M.;Limtrakul,J.J.Mol.Graph.Model. 2006,25,219.
(31) Foresman,J.B.;Frisch,Æ.Exploring Chemistry with Electronic Structure Methods,2nd ed.;Gaussian Inc.:Pittsburgh,PA, 1995;p 64.
(32) Moravetski,V.;Hill,J.R.;Eichler,U.;Cheetham,A.K.;Sauer, J.J.Am.Chem.Soc.1996,18,13015.
(33) Unger,B.;Jancke,H.;Hahnert,M.;Stade,H.J.Sol-Gel Sci. Technol.1994,2,51.
(34) Miertus,S.;Scrocco,E.;Tomasi,J.Chem.Phys.1981,55,117.
(35) Miertus,S.;Tomasi,J.Chem.Phys.1982,65,239.
(36) Cossi,M.;Barone,V.;Cammi,R.;Tomasi,J.Chem.Phys.Lett. 1996,255,327.
(37) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.02;Gaussian Inc.:Wallingford,CT,2004.
(38)Ahlrichs,R.;Bär,M.;Häser,M.;Horn,H.;Kölmel,C.Chem. Phys.Lett.1989,162,165.
(39) Lercher,J.A.;Rumplmayr,G.Appl.Catal.1986,25,215.
(40)Long,L.H.;Wan,Y.B.;Fu,Z.H.;Zhu,H.Y.;Huang,D.P. Industrial Catalysis 2004,12,11.[龙立华,万炎波,伏再辉,朱华元,黄道培.工业催化,2004,12,11.]
(41)Ke,M.;Wang,X.Q.;Zhang,F.M.Petrochemical Technology 2005,34,226.[柯 明,汪燮卿,张凤美.石油化工,2005, 34,226.]
(42)Mora-Fonz,M.J.;Catlow,C.R.A.;Lewis,D.W.Angew.Chem. Int.Edit.2005,44,3082.
(43) Gonzales,N.O.;Chakraborty,A.K.;Bell,A.T.Catal.Lett. 1998,50,135.
(44) Barbosa,L.A.M.M.;van Santen,R.A.J.Phys.Chem.B 2003, 107,4532.
(45) Wilson,E.B.,Jr.Phys.Rev.1934,45,706.
(46) Chakraborty,B.;Viswanathan,B.Catal.Today 1999,49,253.
(47) Corrsin,L.;Fax,B.J.;Lord,R.C.J.Chem.Phys.1953,21, 1170.
April 29,2011;Revised:May 18,2011;Published on Web:June 3,2011.
Phosphorous Moieties in P-ZSM-5 Zeolites
YANG Jing SUN Ying-Xin ZHAO Li-Feng SUN Huai*
(School of Chemistry and Chemical Engineering,Shanghai Jiao Tong University,Shanghai 200240,P.R.China)
Phosphorus modified ZSM-5(P-ZSM-5)zeolite was investigated using quantum mechanics density functional theory and our own N-layer integrated molecular orbital molecular mechanics method (ONIOM).Extra-framework phosphate and in-framework moieties containing phosphorus were found to be plausible local structures in P-ZSM-5 zeolites based on the calculated heats of formation and the free energy data from the hypothetical reactions.Furthermore,we find that the cationic moieties are favored at room temperature.The in-framework acidic moieties are more stable at high temperatures and the stability of the in-framework phosphorus moieties is insensitive to temperature changes.The calculated27Al,31P, and29Si chemical shifts,acidity changes,and structural parameters agree well with the known experimental observations.
H-ZSM-5 zeolite;P-modification;ONIOM method;Density functional theory
O641
*Corresponding author.Email:huaisun@sjtu.edu.cn;Tel:+86-21-54748987.
The project was supported by the National Natural Science Foundation of China(21073119),National Key Basic Research Program of China(973) (2007CB209701)and Research Program from Shanghai Research Institute of Petrochemical Technology,China.
国家自然科学基金(21073119),国家重点基础研究发展计划(973)(2007CB209701)和上海石油化工研究院资助项目
猜你喜欢
电子乐园·上旬刊(2022年5期)2022-04-09
煤气与热力(2021年9期)2021-11-06
湖南饲料(2021年3期)2021-07-28
中国新技术新产品(2020年5期)2020-05-06
天然气化工—C1化学与化工(2019年6期)2019-02-18
铜仁学院学报(2018年6期)2018-07-05
广州化学(2016年5期)2016-11-15
衡阳师范学院学报(2016年3期)2016-07-10
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
应用化工(2014年1期)2014-08-16
