
皮肤淤斑、血小板减少伴肾功能损害
2011-11-26 01:19:10全军肾脏病研究所学术委员会
肾脏病与透析肾移植杂志 2011年3期
全军肾脏病研究所学术委员会
病例摘要
病史47岁男性患者,因“乏力半月,皮肤淤斑、尿色加深10d”于2010-04-22入院。
2010年4月初因劳累出现乏力,伴双下肢酸痛、左下肢皮肤瘙痒、双侧膝关节虫爬感,活动后好转,未予重视。2010-04-09出现上腹部不适,自服“头孢”及护胃药(各1片),无明显好转。4月12日出现尿色加深(呈浓茶色),无尿频、尿急、尿痛,无血块、血丝和尿量减少;同时出现口腔黏膜血疱,双下肢皮肤散在淤点、淤斑,右眼球结膜出血,皮肤巩膜黄染;无发热、腰痛、眼眶痛等不适。次日当地住院测血压170/100 mmHg;尿沉渣红细胞计数256个/μl(正常 <50 个/μl)、蛋白 2+,尿素氮(BUN)18.52 mmol/l、血清肌酐(SCr)280 μmol/L,胆红素升高、间接胆红素为主(总胆红素142.7μmol/L、间接胆红素128μmol/L),血清酶学指标升高(谷草转氨酶78 U/L、乳酸脱氢酶2 608 U/L、羟丁酸脱氢酶1 890 U/L、肌酸激酶416 U/L),血小板极度减低(1×109~20×109/L),贫血进行性加重[血红蛋白(Hb)109 g/L降至71 g/L];上腹部CT未见明显异常,自诉骨髓细胞检查提示“特发性血小板减少性紫癜”;予甲泼尼龙冲击治疗(总量1g),输注血小板及其对症支持治疗。4月19日Hb降至65 g/L,肾功能进一步恶化(BUN 45.4 mmol/l,SCr 458.2 μmol/L),酶学指标进一步升高(乳酸脱氢酶3 191 U/L,羟丁酸脱氢酶2 488 U/L),空腹血糖6.79 mmol/L;溶血检查:游离血红蛋白25mg%,Ham's试验、Coomb's试验、G6PD荧光斑点试验、PK荧光斑点试验均阴性。4月20日出现颜面及双下肢水肿,活动后心慌、气促,夜间不能平卧,伴头晕,尿量减少,急诊收入病房。4月初曾将右手小拇指划伤,愈合欠佳。起病初曾有短暂头痛,自行好转,无行为改变、言语困难;病程中无腹泻、恶心、呕吐,无发热、关节痛,无口腔溃疡、脱发、光过敏、面部红斑。患病以来睡眠、饮食、精神差,大便秘结,体重无明显变化。
既往史 平素常有胃部不适,1997年曾于饮酒后出现“胃出血”;发现血压偏高6年,波动于140/90 mmHg左右,未服用药物治疗。
个人史 从事运输业,2003年~2007年在硫酸化工厂从事机修工作。无长期服药史。
家族史无特殊。
体格检查 体温36.5℃,脉搏88次/min,呼吸16次/min,血压 141/92 mmHg,神志清楚,急性病容,贫血貌,颜面水肿,胸前及四肢皮肤散在淤点、淤斑,巩膜皮肤无明显黄染。全身淋巴结无明显肿大;颈静脉充盈,无怒张;咽不红,扁桃体不大。心界不大,心率88次/min,律齐,各瓣膜区未闻及明显杂音;双肺呼吸音清,未及明显干湿啰音;腹部平坦,肝脾未及,无压痛、反跳痛,移动性浊音阴性。四肢关节活动良好,双下肢轻度水肿。神经系统查体未见异常。
实验室检查
尿液检查 尿沉渣红细胞720万/ml(80%多形型,20%均一),尿蛋白定量3.56 g/24h;C3 10.0mg/L(正常≤2.76mg/L),α2巨球蛋白 3.2mg/L(正常 ≤2.87mg/L);尿糖 2+,NAG酶 3.2 u/(g·Cr)[正常≤16.5 u/(g·Cr)],RBP 34.06mg/L(正常≤0.5mg/L),溶菌酶23.75mg/L(正常<1.0mg/L),胱抑素 C 6.7mg/L。钠滤过分数6.14%,肾衰指数8.97。
血常规 Hb 51g/L,红细胞压积0.159,网织红细胞计数 258.7×109/L,占16%,白细胞 10.6×109/L(中性粒细胞 73%,淋巴细胞 21%),血小板38×109/L,C反应蛋白19mg/L。
血生化 总胆红素/间接胆红素 31.2/24.8 μmol/L,白蛋白/球蛋白 31.0/23.4 g/L,谷草转氨酶/谷丙转氨酶 28.4/91.1 U/L,乳酸脱氢酶/肌酸激酶 2728/597 U/L;SCr 612.6μmol/L,BUN 47.5 mmol/L,尿酸 642 μmol/L,胱抑素 C 5.45mg/L;总胆固醇/三酰甘油 4.65/2.5 mmol/L,钠 146.1 mmol/L,钾 3.21 mmol/L,氯 102.7 mmol/L,钙 2.41 mmol/L,磷1.28 mmol/L,总二氧化碳31.3 mmol/L,葡萄糖5.8 mmol/L。
凝血功能 凝血酶原时间11.3s,国际标准化比值0.98,活化的部分凝血酶时间27.1s,凝血酶时间 15.6s,纤维蛋白原 209mg/dl。
免疫功能 免疫球蛋白IgG 7.59 g/L,IgA 1.3 g/L,IgM 0.597 g/L,抗“O”和类风湿因子正常;补体C3 0.353 g/L,C4 0.126 g/L;自身抗体 ANA、A-dsDNA、ENA多肽谱、ANCA均阴性。CD4+细胞169个/μl(651.3 ±273.6 个/μl),CD8+细胞 123个/μl(452.62 ± 210.83 个/μl),CD4+/CD8+1.38(1.56±0.58),CD20+细胞 123 个/μl(125.22 ±51.55 个/μl)。
溶血试验 Coomb's试验阴性,血酸化溶血试验阴性,血清结合珠蛋白测定44.9 mgHb%(正常38~126 mgHb%),游离血红蛋白2.0mg%(正常0~5mg%);外周血红细胞碎片>50枚/片。
内皮细胞功能 黏附分子VCAM 1 536.77 ng/ml(正常300~1 000 ng/ml),循环内皮细胞25个/ml(正常<20个/ml),血管性血友病因子(vWF)196.51%(正常<200%),血栓调节蛋白 thrombomodulin 14.32 ng/ml(正常 3.15~5.8 ng/ml),血 清 选择素(E-selection)61.16(正常35~65)。
C3肾炎因子、H因子均正常。
(4月30日 外院)血浆ADAMTS 13活性0%,血浆ADAMTS 13抑制物阳性。
辅助检查 胸片:双上肺陈旧性结核。双肾B超:左肾122 mm×54 mm×61 mm,右肾112 mm×54 mm×57 mm,皮质厚度不清,回声增强,皮髓界限欠清晰,集合系统正常。肝胆胰脾超声:肝脏弥漫性病变,胆囊内胆泥形成,胰脾声像图未见占位。心脏彩超:主动脉窦部扩张,左室舒张功能减低,轻度二尖瓣及主动脉瓣返流,少量心包积液。头颅MRI:头颅平扫未见异常,左侧上颌窦炎症。心电图 :窦性心律,左心室高电压,ST-T改变(ST:V5、V6↓≤0.05 mv,T:I、avL、II、V5 偏低)。
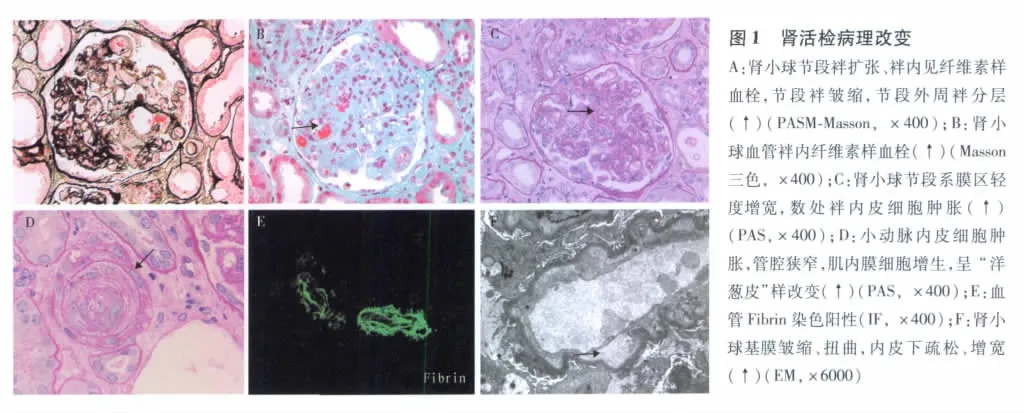
肾脏病理(2010-05-06)
光镜 37个肾小球中2个肾小球袢皱缩明显,余肾小球节段袢扩张,节段袢皱缩,袢内见透明血栓和纤维素样血栓(图1A、B),单个核及中性粒细胞3~10个/球,节段系膜区轻度增宽,数处袢内皮细胞肿胀(图1C)。PASM-Masson:节段外周袢分层(图1A)。多灶性肾小管上皮细胞扁平、刷状缘脱落,散在上皮细胞粗颗粒变性,偶见细胞崩解,管腔见蛋白管型和红细胞管型,偶见嗜碱性物和磷酸钙和草酸钙结晶,偏振光下有折光性,数处灶性肾小管萎缩、基膜增厚,间质较多单个核细胞浸润。小动脉内皮细胞肿胀,管腔狭窄,多处纤维素样坏死,节段内皮下疏松,肌内膜细胞增生,个别呈葱皮样改变(图1D)。
免疫荧光 肾小球、小管阴性,动脉血管Fibrin阳性。(图1E)
电镜 观察2个肾小球,外周袢多数皱缩、扭曲,内皮下疏松、增宽,并见单核细胞浸润至内皮下,致袢腔狭小,内皮下、上皮侧无电子致密物分布(图1F)。足细胞胞质较多微绒毛化,足突广泛融合70~80%。间质水肿,多处近端小管刷状缘及胞质脱落,部分小管上皮细胞出现细胞核固缩。间质未见小动脉。
诊断分析
本例为中年男性患者,起病急、病程短、进展迅速,主要临床表现为血液系统及肾脏受累。血液系统表现为血小板减少和进行性贫血,辅助检查证实血清乳酸脱氢酶、间接胆红素升高,网织红细胞升高,符合溶血性贫血特点;但溶血相关的免疫学检查均为阴性,不支持免疫免疫性溶血性贫血;外周血红细胞碎片阳性,可以明确为微血管病性溶血性贫血(MAHA)[1]。结合患者有高血压和肾脏损害的特点,其临床诊断首先考虑溶血尿毒综合征/血栓性血小板减少性紫癜(HUS/TTP)。
HUS和TTP病理改变均为血栓性微血管病(thrombotic microangiopathy,TMA),其根本原因是内皮细胞损伤和异常的血小板聚集。HUS和TTP临床表现多有重叠,成年患者的治疗也极为相似,有学者认为它们是一种疾病的两种不同临床表现,为不失时机尽早治疗以争取最佳疗效,临床上常不作严格的区分,将其统称为HUS/TTP。但两者的流行病学、临床经过和预后均有差异(表1),发病机制亦不同,临床需进一步鉴别、分型,寻找病因,以便针对性治疗。HUS分为典型和非典型两大类,典型HUS常于产志贺毒素细菌感染后发生,预后良好;非典型HUS多由补体因子异常所致,预后较差。在TTP中,vWF裂解酶ADAMTS13活性严重缺乏(<5%)是导致血小板血栓形成的主要原因。为明确诊断,本例患者入院后即留取血标本送外院查血浆ADAMTS 13活性(残余胶原结合法),发现酶活性严重缺乏且抑制物阳性;血浆补体C3水平偏低,但C3肾炎因子和H因子正常,同时也可以排除其他继发TMA病因[2],故出院诊断为特发性血栓性血小板减少性紫癜。
治疗及随访
入院后给予血浆治疗和大剂量甲泼尼龙冲击(累计5g)治疗。住院期间共行血浆置换7次(1 700~2 000 ml/次),间断输注冰冻血浆及冷沉淀上清液,血浆治疗总量达14 420 ml(197.5 ml/kg),同时积极给予连续肾脏替代治疗及对症支持。经上述治疗后,患者血小板计数逐渐上升、乳酸脱氢酶下降、血色素稳定,复查血浆ADAMTS 13活性恢复正常(100%),抑制物阴性,血小板计数再度上升(图2)。出院时患者尿量正常,SCr降至 229.8 μmol/L,血小板波动于70~92×109/L,乳酸脱氢酶降至250 U/L,Hb稳定于100g/L左右。出院后继续口服泼尼松30mg/d,辅以复方α酮酸、新肾炎胶囊、百令胶囊等保肾,促细胞生成素、铁剂、叶酸等纠正贫血以及利尿、降糖、护胃等对症支持治疗。

图2 主要治疗及病情变化

表1 HUS与TTP的鉴别诊断
6月18日我院门诊复查血小板降至51×109/L,乳酸脱氢酶升至 820 U/L,SCr 247.52 μmol/L,Hb 105 g/L,白蛋白 32 g/L;尿蛋白定量5.2 g/24h(尿量1 760 ml/24h)、尿沉渣红细胞计数235万/ml,考虑病情复发;于外院血浆置换三次并输注3次长春新碱,血小板持续降低,肾功能稳定。7月复查血小板升高、ADAMTS13活性正常,泼尼松逐渐减量。8月出现带状疱疹,神经疼痛明显,饮食、睡眠差,贫血加重,Hb波动于40~50 g/L,血小板正常,予间断输注红悬、血浆,抗病毒等对症支持,带状疱疹逐渐缓解。10月复查血小板51×109/L,Hb 67 g/L,SCr 389.0 μmol/L,BUN 22.42 mmol/L,尿酸678μmol/L,白蛋白 33.5g/L,肝酶正常,乳酸脱氢酶未查;尿蛋白定量1.01 g/24h(尿量900 ml/24h)、尿沉渣红细胞计数 9万/ml;血浆补体C3/C4 0.654/0.173 g/L。因肾功能损害进展,12月开始维持性肾脏替代治疗。
讨 论
血栓性血小板减少性紫癜(TTP)的诊断 TTP由Moschcowitz于1925年首先报道,1958年由Singer等命名,Amorosi和Utman进一步归纳其临床五大特征:发热、血小板减少、MAHA、神经系统症状和肾脏损害,称为“五联征”。多数人认为根据“三联征”(血小板减少,MAHA,神经系统症状)即可诊断TTP,但也有认为必须具备“五联征”才能诊断。
目前常用的诊断标准如下[3]:(1)主要表现:(a)溶血性贫血:末梢血涂片可见裂红细胞;(b)血小板减少:<100×109/L。(2)次要标准:(a)发热(体温超过38.3℃);(b)特征性的神经系统症状;(c)肾脏损害,包括SCr>177μmol/L(2mg/dl)及(或)尿检发现血尿、蛋白尿和(或)管型尿。若有两个主要表现加上任何一个次要标准,诊断即可成立。此例患者满足两条主要表现及一个次要标准,TTP临床诊断成立。
TTP分为先天性和获得性两类[4]。先天性即家族性或复发性,多表现为规律性和间歇性(3~4周)发作;获得性又分为:急性特发性(起病快,散发性,常单次发作),间歇性(病程迁延,反复发作,发作间歇期为数月至数年),继发性(有特定病因可寻者)。
TTP是临床诊断,并无特异指标或金标准,血小板减少与MAHA“二联征”是最为敏感、最具普遍意义、最易引人注意且必不可少的两项诊断依据,可见于100%的患者,而神经系统异常(63%)、肾脏异常(59%)、发热(24%)等的出现率均较低。过去一度强调“三联征”、“五联征”在诊断TTP中的敏感性与特异性,但目前认为到此阶段已进入疾病较晚期,不利于疾病的早期诊治。与以往相比,通过“二联征”确定诊断的标准更为宽泛。值得注意的是,个别患者在病初甚至不具备“二联征”中的任意一项,但随着病情的进展,可迅速表现出来,因此密切的临床观察与不失时机的反复化验检查实属必需。
在确定MAHA时,除溶血性贫血的相关表现外,在外周血发现裂红细胞(红细胞碎片)是极为重要的根据。过去曾以裂红细胞>2%为诊断依据之一,但临床上一些非TMA的情况也可出现较多裂红细胞,必须仔细甄别。TTP时外周血裂红细胞的出现时间可能晚于血小板减少与溶血,故应把握好检查时机,且其数量多少与病变程度有关。以往曾报道过裂红细胞始终阴性的患者[5,6],因而近年的文献中已很少规定诊断时所需的裂红细胞数量标准。
TTP患者的神经精神异常表现常有一过性、反复性、多样性与多变性等特征,起病早期易被忽视,且局灶性损伤的表现少见,CT/MRI等影像学检查多无特殊发现。患者起病初期曾出现短暂性头痛,不能排除其与病情相关。
镜下血尿和非肾病性蛋白尿是TTP患者最常见的尿检异常。国外一项回顾性研究发现216例TTP患者有78%出现血尿,75%出现蛋白尿,另分别有31%和24%出现白细胞尿和管型,肉眼血尿少见[7]。全军肾脏病研究所归纳了27例TMA患者的临床病例特点[8],发现只有2例患者出现肉眼血尿,其余均为镜下血尿;尿蛋白定量不多,仅 3例>3 g/24h。有研究分析23例HUS患儿尿常规发现17/23尿蛋白定性 3+以上,12/23隐血 3+以上[9]。
该患者病程中补体C3水平持续降低,C4正常,提示持续存在补体旁路途径过度活化,且肾脏损害重,临床难以除外补体异常或补体因子异常导致的非典型HUS。但肾组织病理未见明确免疫复合物及补体成分沉积,不支持补体活化导致肾脏损伤;筛查C3肾炎因子及补体H因子均正常,补体C3与TMA病变之间的关系仍无法明确,不能排除存在其他原因导致补体C3降低。TTP患者是否与非典型HUS患者一样可以存在补体调节异常目前还不得而知,有待进一步深入研究。
ADAMTS13活性的检测及意义 在流动血液剪切力作用下,ADAMTS 13作用于vWF A2区842酪氨酸-843蛋氨酸间的肽键,将vWF多聚体裂解为大小不等的小分子肽段,在生理状态下调控vWF的结构与功能。当ADAMTS13活性降低而无法切割超大分子vW因子(ULvWF)时,ULvWF可网罗血浆中的血小板而导致微血管内富含血小板血栓形成[10]。TTP患者体内可缺乏该酶活性,先天性TTP患者主要是因为ADAMTS13基因突变导致酶活性缺失,而获得性TTP患者ADAMTS 13活性缺乏主要是因为消耗过多,其中多数存在针对ADAMTS 13的抗体或抑制物[11]。
Peyvandi等[12]分析了 100例 TTP患者血浆ADAMTS 13活性水平,48例严重缺乏(<10%),28例正常(>46%),87%严重缺乏者检出中和抗体。秦燕等[13]研究表明90%特发性TTP患者ADAMTS 13活性较正常对照极度降低(<5%),83.3%的ADAMTS 13缺乏者可检出抑制物。而继发性TTP和HUSADAMTS 13缺乏罕见,据俄罗斯TTP-HUS登记处统计,92例继发性HUS/TTP病例中无一例出现ADAMTS 13活性缺乏[14]。但ADAMTS 13活性降低除见于TTP患者,也可见于恶性肿瘤发生转移的患者,可不伴TTP的临床表现[15]。此外,研究还发现并非所有先天性ADAMTS 13缺陷患者都会发病,部分患者可在妊娠期发病[16]。可见ADAMTS 13的缺陷参与了TTP的发病过程,但不一定是引起TTP的决定性因素。但ADAMTS 13活性严重缺乏多见于特发性TTP,且多可检出抑制物。
ADAMTS 13活性不仅对于诊断TTP有相当的敏感性(33%~100%)和特异性,而且有助预后判断与治疗方法选择。严重缺乏(<5%)多为特发性TTP的特异表现,对血浆置换反应良好,ADAMTS 13正常者则预后差,继发性TTP的ADAMTS 13常可正常。但ADAMTS 13活性测定有多种方法,检测结果的判读标准尚有待统一,且其应用历史尚短,还需要继续积累经验,因此很多指南未将其列为主要诊断依据。该患者进行了多次ADAMTS13活性检测,其中仅有一次严重缺乏,是否存在检验误差或结果判读错误不得而知。
小结:对于临床表现为HUS/TTP的患者,应深入挖掘病史,寻找对鉴别诊断有意义的线索,同时还要尽可能利用已开展的实验室检测项目了解患者存在的病理生理变化,为进一步鉴别疾病类型或亚型提供有力证据。TTP起病急,病程进展迅速,有致命危险,一旦诊断应尽快治疗。血浆置换治疗不仅能够补充血浆中缺乏的ADAMTS 13酶以及凝血所需的vWF,清除ADAMTS 13的抑制剂,而且能去除导致内皮损伤和血小板聚集的细胞因子或自身抗体,因而可有效缓解症状。TTP容易复发,维持治疗、预防复发是改善临床预后的保证,但目前仍无有效的维持治疗方法。
1 汪声恒.血栓性血小板减少性紫癜/溶血尿毒综合征.//张之南,沈悌..血液病诊断及疗效标准.北京:科学出版社,2001,P176-181.
2 李世军,刘志红.血栓性微血管病与肾脏损害.//黎磊石,刘志红.中国肾脏病学.北京:人民军医出版社,2008,P573-591.
3 钱 樱,沈志祥.血栓性血小板减少性紫癜.//达万明,裴雪涛.现代血液病学.北京:人民军医出版社,2003,P654-660.
4 Allford SL,Hunt BJ,Rose P,et al.Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias.Br JHaematol,2003,120(4):556 -573.
5 Fava S,Galizia AC.Thrombotic thrombocytopenic purpura-like syndrome in the absence of schistocytes.Br JHaematol,1995,89(3):643-644.
6 Daram SR,Philipneri M,Puri N,et al.Thrombotic thrombocytopenic purpura without schistocytes on the peripheral blood smear.South Med J,2005,98(3):392 -395.
7 Eknoyan G,Riggs SA.Renal involvement in patients with thrombotic thrombocytopenic purpura.Am J Nephrol,1986,6(2):117 - 131.
8 李世军,刘志红,陈惠萍,等.血栓性微血管病的肾脏损害——附27例临床病理分析.肾脏病与透析肾移植杂志,2006,15(5):428-433.
9 叶礼燕,姜采荣,余自华,等.溶血尿毒综合征23例诊治分析.临床儿科杂志,2008,17(8):690 -693.
10 Fujikawa K,Suzuki H,McMullen B,et al.Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family.Blood,2001,98(6):1662-1666.
11 Zheng XL,Kaufman RM,Goodnough LT,et al.Effect of plasma exchange on plasma ADAMTS 13 metalloprotease activity,inhibitor level,and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura.Blood,2004,103(11):4043-4049.
12 Peyvandi F,Ferrari S,Lavoretano S,et al.von Willebrand factor cleaving protease(ADAMTS-13)and ADAMTS-13 neutralizing autoantibodies in 100 patients with thrombotic thrombocytopenic purpura.Br JHaematol,2004,127(4):433 -439.
13 秦 燕,苏 健,黄石兵,等.ADAMTS13活性及抑制物测定在ITTP诊断中的价值.南通医学院学报,2009,29(6):437-438.
14 Bohm M,Betz C,Miesbach W,et al.The course of ADAMTS-13 activity and inhibitor titre in the treatment of thrombotic thrombocytopenic purpura with plasma exchange and vincristine.Br J Haematol,2005,129(5):644 -652.
15 Oleksowicz L,Bhagwati N,Deleun-Fernandez M.Deficient activity of vonwillebrand's factor-cleaving p rotease in patients with disseminated malignancies.Cancer Res,1999,59(9):2244 -2250.
16 Furlan M,Robles R,Galbusera M,et al.von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolyticuremic syndrome.N Engl J Med,1998,339(22):1578 -1584.
猜你喜欢
家教世界(2022年34期)2023-01-08 13:52:50
昆明医科大学学报(2021年8期)2021-08-13 09:00:02
昆明医科大学学报(2021年2期)2021-03-29 07:42:24
心肺血管病杂志(2018年11期)2018-12-18 01:51:40
中国医药指南(2017年3期)2017-11-13 02:55:32
兽医导刊(2016年6期)2016-05-17 03:50:31
中国医药生物技术(2015年4期)2015-12-26 08:26:36
实用肝脏病杂志(2015年5期)2015-12-03 06:28:07
河南医学研究(2014年3期)2014-02-27 14:51:59
当代畜禽养殖业(2014年12期)2014-02-27 08:00:10
