
A Novel Synthesis of Fexofenadine Pyridinium Salts by Oxidation of Piperidine Derivatives under Simple Conditions
2011-11-23 01:26:26LIXiaolingSHENChaoZHENGHuiZHANGPengfei
杭州师范大学学报(自然科学版) 2011年5期
LI Xiao-ling,SHEN Chao,ZHENG Hui,ZHANG Peng-fei*
(1.College of Material,Chemistry and Chemical Engineering,Hangzhou Normal University,Hangzhou 310036,China; 2.Hangzhou Great Forest Biomedical Ltd., Hangzhou 310028,China)
A Novel Synthesis of Fexofenadine Pyridinium Salts by Oxidation of Piperidine Derivatives under Simple Conditions
LI Xiao-ling1,2,SHEN Chao1,ZHENG Hui1,ZHANG Peng-fei1*
(1.College of Material,Chemistry and Chemical Engineering,Hangzhou Normal University,Hangzhou 310036,China; 2.Hangzhou Great Forest Biomedical Ltd., Hangzhou 310028,China)
The paper reported an efficient synthetic procedure,which has mild response conditions and high productivity,for the preparation of some novel pyridinium salts by oxidation of piperidine derivatives only using some simple oxidants in high yield at ambient temperature and researches on its response mechanisms.
Pyridinium salts; fexofenadine; piperidine derivatives; oxidation; mechanism
1 Introduction
As one kind of pyridine derivatives,the pyridinium salts are employed as valuable reagents in organic synthesis and in the construction of novel material[1-2].The wide range of chemical and physical properties that the pyridinium salts exhibit have drawn the attention of chemists[3-5].These pyridinium salts are well known for their diverse biological properties,such as antimicrobial[6-7],antifungal[8],antiproliferative activities[9].In addition,some of these pyridinium salts have been proved to be efficient in vitro activators of three carbonic anhydrase isozymes[10].In 2006,several new classes of efficient pyridinium salts were synthesized and tested as gene delivery and the quantitative structural-activity relationship (QSAR) analyses identified the most efficient structural variables for the new gene transfer agents were carried out by Balaban et al[11-12].There has been an upsurge of interest in pyridinium salts as ionic liquids (ILs) which have been studied recently as potential “green” solvents[13-15].Therefore,studies of the synthesis and application of novel pyridinium salts have attracted a great deal of interest in recent years.
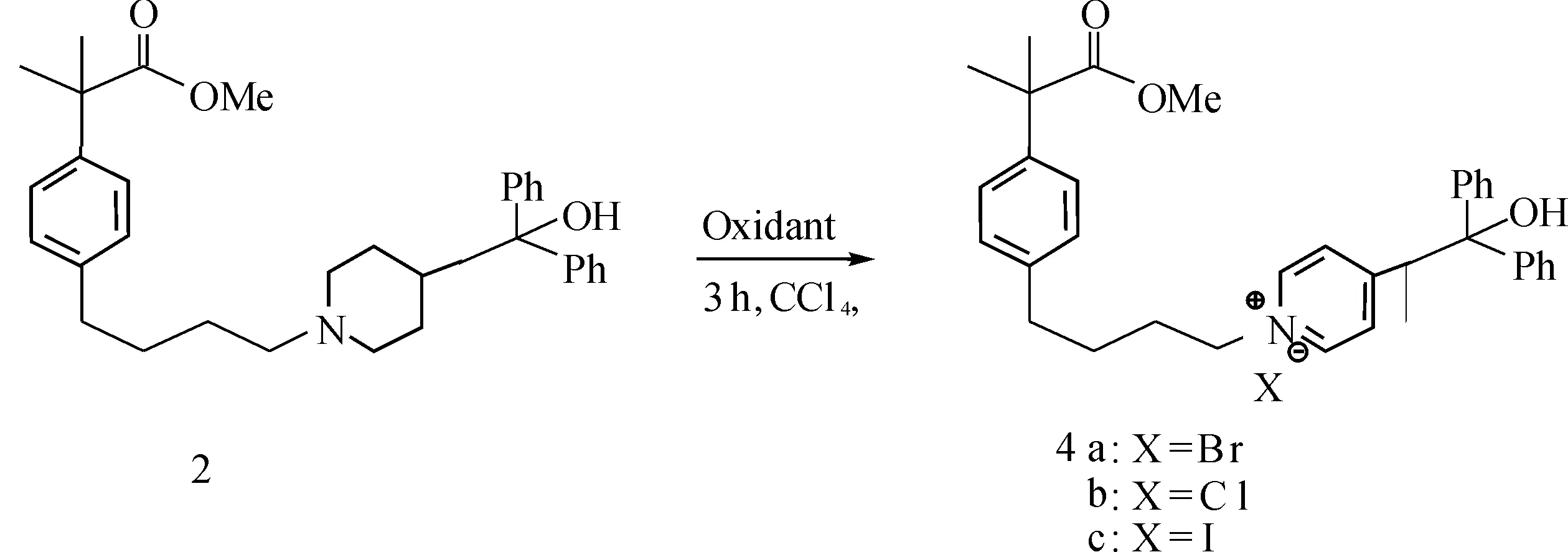
In last three decades,there were serveral types of methodologies that could give access to substituented pyridinium salts.Leonard and coworker reported that some pyridinium salts can be prepared in some oxidation conditions through the use of tertiary amines and mercuric acetate[16].Similarly,a reduced Molybdenum oxide-Alumina catalyst was used in dehydrogenation of piperidine derivatives and the kinetics of the piperidine dehydrogenation was discussed[17].In 1991,some quaternary ammonium salts were synthesized in the vapor-phase reaction over a nickel-containing catalyst by Endo et al[18].Indeed,up to now,the method of catalytic dehydrogenation of piperidine ring always focus on the use of the various heavy metal catalysts composed of Ni,Cr or Pd and these methods needed for high temperature.Recently,the Zincke synthesis of pyridinium salts starting from amino acids was successfully carried out at ambient temperature by Marazano’ group[19],but the process is often tedious and the yield is medorate.Herein,we report a novel synthetic route of pyridinium salts under mild condition in high yields (Scheme 1).

Scheme 1 Synthesis of the unexpected pyridinium salts 4
2 Result and discussion
As part of our ongoing work on the synthesis of fexofenadine,which is an active metabolite of terfenadine in the treatment of seasonal allergic rhinitis and chronic idiopathic urticaria as a first-line therapeutic agent[20-22],a fexible synthetic route for intermediate 3 was designed and shown inScheme1.We believed that this possible synthetic route seems to enable product 3 to be prepared efficiently.The piperidine derivative 1 was readily prepared by reaction of commercially available azacyclonol hydrochloride with corresponding mesylate[23].So the piperidine derivatives 1 was reduced (H2/Pd-C) to give the corresponding intermediate 2 at first and then was treated with halogenating reagents such as NBS,NCS and NIS.We considered the corresponding alkyl halide 3 could be obtained in this condition.However,the chemical and spectral properies of the product did not show agreement with the structure of target compound 3.By analysis of IR and NMR,we found that the piperidine ring was oxidated to give the unexpected pyridinium salts 4.Further evidence comes from the analysis of HRMS of the sample,which shows that the piperidine ring is fully oxidated to a pyridine ring.To the best of our knowledge,this is the first example of pyridine ring prepared in such a approach.It is noteworthy that the reaction condition is simple,the transformation time is short and the yield is high.
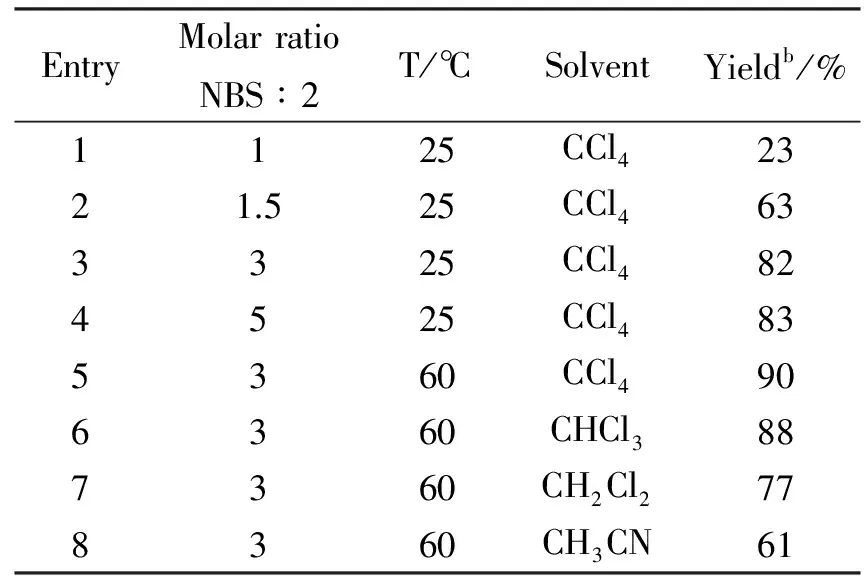
Tab.1 Optimization of oxidative conditionsa
aReaction conditions:Compoun 2(1 mmol),NBS,sovent(8 mL) at corresponding temperature with stirring for 3 h.
bIsolated yields.
To begin the search for more effective conditions for the synthesis of pyridinium salts,we employed intermediate 2 as a model substrate and NBS as the oxidant in CCl4at room temperature.We first optimized the molar ratios of oxidant to substrate.Poor yields were obtained when the molar ratio was 1 (Table1,entry1).The yields could be improved from 63% to 82% when the molar ratio was added from 1.5 to 3.However,much more oxidant did not effect the yield.Therefore,we thought the most suitable molar ratio was 3.The starting materials disappeared in 3 hr monitored by TLC.Some increase in yield was occurred when the temperature was changed from 25 to 60 ℃(entry4 vsentry5).Next,the effect of solvents on this reaction was investigated.When the reaction was carried out in CCl4,the yield was obtained higher than CHCl3,CH2Cl2,and CH3CN (entry5 vsentries6,7,and8).
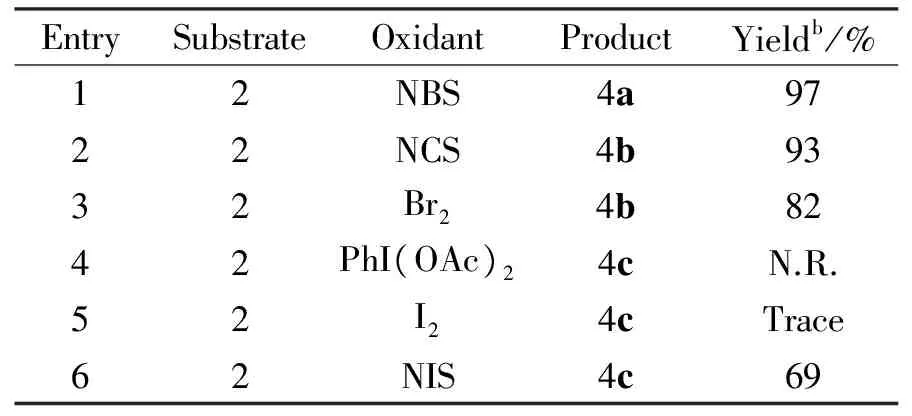
Tab.2 Preparation of pyridinium salts 4a-c from piperidine derivatives 2 by different oxidantsa
aReaction conditions:Compound2(1 mmol),Oxidant(3 mmol),CCl4(8 mL) at 60℃ with stirring for 3 h.
bIsolated yields.
In order to ascertain whether aromatization of piperidine ring might have been favored by the presence of NBS,we selected several other oxidants for reation with the piperidine derivatives 2 in the optimized conditions(Scheme2).Reaction of 2 with NCS,Br2or NIS in 1∶3 molar ratio in CCl4at 60 ℃ for 3h led to the formation of the quaternary salt in 93%,82% and 69% isolated yields(Table2,entries2,3,and6).Whereas none of the desired product was observed when the PhI(OAc)2was used (Table2,entry4),a trace amount of the product was observed with the I2(Table2,entry5).
Scheme 2 Common synthetic procedure
We were disappointed to find that attempts at oxidative dehydrogenation of several other piperidine molecules (including 4-hydroxy-N-methylpiperidine,4-amino-1-benzylpiperidine,4-chloro-N-methylpiperidine,and 1-phenethyl-4-piperd-one) lacking more electron-withdrawing substituents resulted in failure.We reasoned that the presence of hydroxydiphenylmethyl as a electron-withdrawing group is necessary in the reaction system.Oxidative dehydrogenation of piperidine derivatives resulted in a complex mixture of decomposition intermediates.This suggested that the mechanisms and pathways of the oxidation were very complex,involving many chemical reactions as well as a physical mass transfer.
Similar formation of pyridinium salts can easily be prepared by treatment of substrates with N-bromosuccinimide according to some earlier observations and some mechanisms of iminium salts in which the six-membered heterocyclic rings were aromatized have been presented[24-25].However,there was not a probable mechanism to meet our present work.Here,we considered a plausible mechanism for pyridinium salts formation (Scheme3).Initially,attack of the basic nitrogen by the “positive” bromine to form the N-bromo-quarternary complexAwas postulated as the firt step.Then an elimination step involving the tertiary proton of complexAto form the iminium saltsBand bromination gave the intermediateDwhich undergoes elimination to form the complexE.Analogously,the process of “bromination-elimination” occurred again to give the intermediateHwhich could be oxidized by NBS to the target product 4a.Some experimental facts are in agreement with this proposed mechanism,such as the highest isolation yield was obtained in experiments when the oxidizing agent was used in triple substoichiometric amounts.The product 4ashowed the HRMS spectral data which clearly indicated that the six-membered heterocyclic ring was aromatic.

Scheme 3 Possible mechanism for the formation of pyridinium salts 4a
3 Conclusion
In summary,we described here an unexpected and interesting oxidative transformation of piperidine derivatives to pyridine ring by using simple oxidative reagents in easy one-pot synthetic operations.The advantages of this new method are operational simplicity and good yields.Further expansion of the reaction scope and synthetic applications of this methodology are in progress in our laboratory.
4 Experimental section
4.1 General information
All chemicals are commercially available and were purchased in reagent-grade quality; CH2Cl2was distilled from CaH2freshly prior to use; Carbon tetrachloride was also distilled from calcium hydride.Column chromatography was performed on silical gel,Merck grade 60 (230-400mesh).Reactions were monitored by thin layer chromatography (TLC) which was performed on a Merck precoated TLC (silica gel 60 F254) plate.Melting points were determined on an X4-Data microscopic melting point apparatus; IR spectra were determined on a Nicolet NEXUS-470 FT-IR spectrometer as KBr pellets.The1H and13C NMR spectra were recorded in CDCl3on a Bruker AVANCE DRX-400 NMR spectrometer,using TMS as the internal standard.High resolution mass spectra were recorded on a 4.7 Tesla IonSpec ESI-FTMS.4-[4-[4-(Hydroxydiphenylmethyl)-1-piperidinyl]-butyl]-α,α-dimethyl benzeneacetic acid,methyl ester was prepared by a laborious procedure[23].
4.2 General procedure for the synthesis of 4-[ 4-[4-(Hydroxydiphenylmethyl)-1-piperidinyl]-butyl]-α,α-dimethylbenzeneacetic acid,methyl ester (2)
A solution of 4-[4-[4-Hydroxydiphenylmethyl]-l-piperidinyl]-l-butynyll-α,α-dimethylbenzeneacetic acid,methyl ester 1(750 mg,1.48 mmol) in 30 mL ethanol and 0.38 g of Pd/C were added to a 300 mL autoclave under nitrogen atmosphere,and the mixture was stirred at room temperature.Hydrogen was initially introduced into the autoclave at a pressure of 0.2 Mpa before being reduced to 1 atm by carefully releasing the stop valve.After this procedure was repeated three times,the vessel was pressurized to 0.2 Mpa.The reaction mixture was stirred under 0.02 Mpa pressure of hydrogen at 30℃ for 10 h before releasing the hydrogen,and then filtered.The filtrate was concentrated to give 695 mg product in 92% yield;1H NMR (400 MHz,CDCl3)δ=7.47(d,J=8 Hz,4H),7.29~7.09(m,10H),6.40(q,1H),3.63(s,3H),2.96(m,1H),2.93(m,1H),2.57(t,J=9.2 Hz,2H),2.42(m,2H),2.29(t,J=4.4 Hz,2H),1.91(q,2H),1.57(s,6H),1.52(s,8H);13C NMR (100 MHz,CDCl3):δ=177.2,145.9,141.6,140.6,128.2,128.1,127.9,126.6,125.7,79.2,58.6,53.9,51.9,46.2,45.9,35.1,29.2,26.4,26.1,26.0 ppm; IR(KBr):3 442,2 951,1 731,1 259,1 146,1 067,703 cm-1; HRMS:m/z calcd for C33H41O3N [M+Na]+522.298 4,found 522.298 6.
4.3 General Procedure for the Preparation of pyridinium salts (4)
A solution of 4-[ 4-[4-(Hydroxydiphenylmethyl)-1-piperidinyl]-butyl] -α,α-dimethylbeneneacetic acid,methyl ester 2 (380 mg,0.77 mmol) and in Carbon tetrachloride (10 mL) at 25℃,after 10 min N-Bromosuccinimide (411 mg,2.31 mmol) was added.The resulting mixture was stirred for 2 h and then quenched with sat.aqueous NaCl.The mixture was extracted with CH2Cl2.The combined organic layer was washed with water,dried over MgSO4,filtered and concentrated undervacuo.Column chromatography on silica gel afford pure pyridinium salts 4a-c.
4a:a poor yellow product,(433 mg,97%),1H NMR(400 MHz,CDCl3):δ=9.0(d,J=10 Hz,2H),7.96(d,J=6.4 Hz,2H),7.32-7.23(m,10H),7.19-7.08(d,J=6.8 Hz,4H),4.77(t,J=12 Hz,2H),3.62(s,3H),2.74(s,1H),2.62(s,2H),1.97(m,2H),1.67(m,2H),1.54(s,6H);13C NMR(100 MHz,CDCl3):δ=177.2,159.1,145.8,140.5,129.2,128.9,128.6,128.4,128.1,126.5,125.8,79.3,58.5,53.7,51.7,35.1,29.6,29.1,28.2 ppm;IR(KBr):3 425,1 670,1 408,1 139,1 017,925,651 cm-1,HRMS:m/z calcd for C33H36O3N [M-Br]+494.268 9 ,found 494.263 9.
4b:HRMS:m/z calcd for C33H36O3N [M-Cl]+494.268 9,found 494.266 6.
4c:HRMS:m/z calcd for C33H36O3N [M-I]+494.268 9,found 494.265 2.
[1] Wang Qifang,Hui Li,Hou Hong,etal.Synthesis of zwitterionic salts of pyridinium-meldrum acid and barbiturate through unique four-component reactions[J].J Comb Chem,2010,12(2):260-265.
[2] Chen L J,Burka L T.Chemical and enzymatic oxidation of furosemide:formation of pyridinium salts[J].Chem Res Toxicol,2007,20(12):1741-1744.
[3] Singh R P,Winter R W,Gard G L,etal.Quaternary salts containing the pentafluorsulfanyl (SF5) group[J].Inorg Chem,2003,42(19):6142-6146.
[4] Lavilla R,Spada A,Bosch J.Oxidative dephosphonylation of 1,4-dihydropyridines and pyridinium salts[J].Org Lett,2000,2:1533-1535.
[5] Donohoe T J,Connolly M J,Walton L.Regioselective nucleophilic addition to pyridinium salts:a new route to substituted dihydropyridones[J].Org Lett,2009,11(23):5562-5565.
[6] Pernak J,Kalewska J,Ksycinska H,etal.Synthesis and anti-microbial activities of some pyridinium salts with alkoxymethyl hydrophobic group[J].Eur J Med Chem,2001,36(11/12):899-907.
[7] Maeda T,Manabe Y,Yamamoto M,etal.Synthesis and antimicrobial characteristics of novel biocydes,4,4’-(1,6-hexamethylenedioxy dicarbonyl)bis (1-alkylpyridinium iode)s[J].Chem Pharm Bull,1999,47(7):1020-1023.
[8] Daniel O,Namfon P,Rosemary H,etal.Synthesis,antifungal,heamolytic and cytotoxic activities of a series of bis(alkylpyridinium)alkanes[J].Bioorg Med Chem,2009,17(17):6329-6339.
[9] Adamec J,Beckert R,Weiβ D,etal.Hybrid molecules of estrone:new compounds with potentia antibacterial,antifungal,and antiproliferative activities[J].Bioorg Med Chem,2007,15(8):2898-2906.
[10] Monica I,Mircea D B,Marc A I,etal.Carbonic anhydrase activators:design of high affinity isozymes Ⅰ,Ⅱ,and Ⅳ activators,incorporating tri-/tetrasubstituted-pyridinium-azole moieties[J].J Med Chem,2002,45(2):504-510.
[11] Marc A I,William A S,Betty H J,etal.Lipophilic pyrylium salts in the synthesis of efficient pyridinium-based cationic lipids,gemini surfactants,and lipophilic oligomers for gene delivery[J].J Med Chem,2006,49(13):3872-3887.
[12] Marc A I,William A,Ion G,etal.Pyridinium cationic lipids in gene delivery:a structure-activity correlation study[J].J Med Chemk,2004,47(15):3744-3754.
[13] Zhu Yinghuai,Carpenter K,Bun C C,etal.(R)-binap-mediated asymmetric hydrogenation with a rhodacarborane catalyst in ionic-liquid media[J].Angew Chem Int Ed,2003,42(32):3792-3795.
[14] Koen B.Lonic liquid crystals[J].Chem Rev,2005,105:4148-4204.
[15] Schuster O,Yang L R,Raubenheimer H G,etal.Beyond conventional N-heterocyclic carbenes:abnormal,remote,and other classes of NHC ligands with reduced heteroatom stabilization[J].Chem Rev,2009,109(8):3445-3478.
[16] Leonard N J,Hay A S,Fulmer R W,etal.Unsaturated amines.Ⅲ.Introduction of α,β-unsaturation by means of mercuric acetate :△1(10)-Dehydroquinolizidinel,2[J].J Am Chem Soc,1956,77:439-444.
[17] Sonnemans J,Janus J M,Mars P.Hydrogenation and paiperidine and dehydrogenation[J].J Phys Chem,1976,80:2107-2110.
[18] Lee S B,Toshikszu T,Takeshi E.Quaternary ammonium salts as useful cationic initiators.6.synthesis,activity,and thermal latency of N-benzylpyridinium salts and the role of the pyridine moiety[J].Macromolecules,1991,24(10):2689-2693.
[19] Tuan M N,Maria R S,Jean-Charles W,etal.Aminopentadiene imines from zincke salts of 3-alkylpyridines.Application to a synthesis of pyridininium salts from amino acids[J].J Org Chem,2007,72(15):5916-5919.
[20] Simpson K,Jarvis B.Fexofenadine hydrochloride:terfenadine carboxylate hydrochloride MDL-16455A Allegra[J].Drugs Future,1996,7:1017-1020.
[21] Reinhold T,Thomas S,Martin P.Syntheses and pharmacological properties of the histaminic H1antagonists sila-terfenadine-A,sila-terfenadine-B,disila-terfenadine,and sila-fexofenadine:a study on C/Si bioisosterism[J].Organometallics,2004,23(21):4915-4923.
[22] Mao Jiangang,Gu Haining,Zhang Pengfei.A novel and efficient synthesis of intermediates for the preparation of fexofenadine[J].Scholarly Research Exchange,2008:13709-13711.
[23] Stephen H K,Robert J H,George J A.Facile synthesis of an oxidation product of terfenadine[J].J Org Chem,1994,59(9):2620-2622.
[24] Rao K V,Kapicak L S.Action of N-bromosuccinimide on some indolizidine and quinolizidine systems[J].J Heterocycl Chem,1976,13(5):1073-1077.
[25] Chuang T H,Lee S J,Yang Chengwei,etal.Expedient synthesis and structure-activity relationships of phenanthroindolizidine and phenanthroquinolizidine alkaloids[J].Org Biomol Chem,2006,4(5):860-867.
氧化法合成非索非那定吡啶盐及其机理研究
李小玲1,2,沈 超1,郑 辉1,章鹏飞1
(1.杭州师范大学材料与化学化工学院,浙江 杭州 310036;2.杭州广林生物医药有限公司,浙江 杭州 310028)
报道了一种用简单氧化剂将哌啶衍生物氧化为相应吡啶盐的方法,该方法具有反应条件温和、产率高等优点,并对反应机理进行了研究探讨.
吡啶盐;非索非那定;哌啶衍生物;氧化;机理
date:2011-02-28
National Natural Science Foundation of China (21076052).
Biography:LI Xiao-ling(1983—),female,born in Wenzhou,Zhejiang province,master,engaged in pharmaceutical synthesis.
*CorrespondingauthorZHANG Peng-fei(1965—),male,born in Jixi,Anhui province,doctor,engaged in pharmaceutical synthesis and asymmetric synthesis.E-mail:chxyzpf@hotmail.com
10.3969/j.issn.1674-232X.2011.05.007
O621.25+4.1ArticlecharacterA
1674-232X(2011)05-0416-06
猜你喜欢
浙江化工(2024年2期)2024-03-15 02:27:40
无机化学学报(2024年1期)2024-01-20 03:55:50
健康体检与管理(2022年2期)2022-04-15 22:33:17
化工职业技术教育(2021年5期)2021-11-09 03:10:40
考试与评价·高一版(2021年3期)2021-08-14 22:49:16
化工职业技术教育(2021年1期)2021-03-15 06:57:44
快乐作文·低年级(2016年7期)2016-09-22 19:07:34
化工学报(2016年3期)2016-03-14 08:37:00
合成化学(2015年1期)2016-01-17 08:53:55
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
