
环状氨基甲酸酯衍生物的合成及反应研究
2011-09-17 00:54王家喜
河北工业大学学报 2011年6期
王 静,郭 爽,王家喜
(河北工业大学 化工学院,天津 300130)
环状胺基甲酸酯类化合物可广泛用作农药、医药、合成树脂改性和有机合成的中间体,其作为有机合成的中间体可用于合成异氰酸酯、杂环化合物、无毒聚氨酯、氨基树脂等,与烯烃、醛、酮发生加成反应可生成多种用途的产品[1-2].传统生产工艺多采用光气为原料[3],由于光气具有剧毒且该工艺中会产生大量腐蚀性气体,存在严重的安全隐患,所以非光气法的胺基甲酸酯制备引起了人们的广泛关注[4].
本论文以“绿色化学品”的碳酸二甲酯与醇胺的反应,合成环状胺基甲酸酯,并探索了环状胺基甲酸酯与胺的反应.
1 实验部分
1.1 试剂与仪器
试剂:乙醇胺、碳酸二甲酯、丙烯酸甲酯、1,6-己二胺、醋酸锌、正丁胺为分析纯,碳酸钾为化学纯,天津大学科威公司.
仪器:AVANCE/400型核磁共振谱仪,瑞士布鲁克公司;SP-6800A型气相色谱仪,山东鲁南瑞虹化工仪器有限公司;N2000色谱数据工作站,浙江大学智能信息工程研究所;XT4-100A熔点仪,北京科仪电光仪器厂;Vector-22型红外光谱仪,德国BRUCK公司.
1.2 合成方法
1.2.1 2-噁唑烷酮(Ⅰ)的合成
在装有温度计、磁子搅拌和冷凝管的三口瓶内依次加入48.86g(0.800mol)乙醇胺、72.06g(0.800mol)DMC和2.4184g已活化的醋酸锌,升温到80℃回流反应4h,脱溶剂后用无水乙醇重结晶,得48.36g产物Ⅰ(收率69.5%),熔点84.0~86.0℃(文献 [5]:86.5~88.5℃).1HNMR(CDCl3,400 MHz, ,ppm):6.32(br,-NH),4.46(t,J=7.2 Hz,2H,-CH2O),3.65(t,J=7.2 Hz,2H,-CH2NH).
1.2.2 2-(2-噁唑烷酮)丙酸甲酯(Ⅱ)的合成
在装有温度计、磁子搅拌和冷凝管的三口瓶内依次加入4.35 g(0.05 mol)Ⅰ、4.30 g(0.05 mol)丙烯酸甲酯和0.0865g的K2CO3,升温到80℃反应2.5h,得到8.65g浅黄色透明溶液产物Ⅱ[6].1HNMR(CDCl3,400MHz, ,ppm):4.32(t,J=7.2Hz,2H,-CH2O),3.70(s,3H,-OCH3),3.63(t,J=7.2 Hz,2H,-CH2CH2),3.58(t,J=6.6 Hz,2H,-CH2N),2.63(t,J=6.6 Hz,-CH2CO).
1.2.3 Ⅱ和1,6-己二胺的反应
在装有温度计、磁子搅拌和冷凝管的三口瓶内依次加入8.65 g(0.050mol)Ⅱ、2.90g(0.025 mol)1,6-己二胺和0.086 5 g的K2CO3,升温到70℃反应5 h,蒸馏反应中生成的甲醇,减压脱溶剂后得到9.84 g微黄色固体产物Ⅲ.1HNMR(CDCl3,400MHz, ,ppm):6.78(br,-NH),4.32(t,J=7.2Hz,2H,-CH2O),3.65(t,J=7.2 Hz,2H,-CH2CH2),3.58(t,J=6.6 Hz,2H,-CH2N),3.22(m,2H,-CH2NH),2.50(m,2H,-CH2CO),1.49(m,2H,-CH2CH2),1.33(m,2H,-CH2CH2).
1.2.4 Ⅲ和1,6-己二胺的反应
在装有温度计、磁子搅拌和冷凝管的三口瓶内依次加入9.95 g(0.025mol)Ⅲ、2.90g(0.025 mol)1,6-己二胺、0.1285g的K2CO3和9 g无水乙醇,在100~110℃反应10h,蒸出乙醇,体系温度逐渐升高到150℃后恒温反应6 h,减压浓缩,得到12.5 g黄色蓬松状略有弹性的固体产物Ⅳ.FT-IR(,cm1):3 301(br),3076(w),2926(s),2854(w),1743(w),1643(s),1562(s),1461(br),1371(w),1270(w),1 121(w),1 051(w),879(w),761(w),727(w),593(w).
1.2.5 Ⅱ和正丁胺的反应
在装有温度计、磁子搅拌及冷凝管的三口瓶内依次加入7.84 g(0.045 mol)Ⅱ、6.62 g(0.091 mol)正丁胺和0.1119g的K2CO3,升温到82℃反应,由滴液漏斗缓慢滴加23.3g正丁胺,不断蒸出副产物甲醇,GC跟踪反应至无甲醇分出.将低沸点的馏分全部蒸出,得到10.51 g浅棕红色溶液产物Ⅴ.1H NMR(CDCl3,400MHz, ,ppm):6.52(s,H,-NH),4.30(t,J=7.2Hz,2H,-CH2O),3.65(m,2H,-CH2CH2),3.55(m,2H,-CH2N),3.24(m,2H,-CH2NH),2.49(t,J=6.4 Hz,2H,-CH2CO),1.48(t,J=7.6 Hz,2H,-CH2CH2),1.35(t,J=7.6 Hz,2H,-CH2CH2),0.93(m,3H,-CH2CH3).
1.2.6 Ⅴ和1,6-己二胺的反应
在装有温度计、磁子搅拌及冷凝管的三口瓶内依次加入6.40 g(0.030mol)Ⅴ、1.72g(0.015 mol)1,6-己二胺、0.081 4 gK2CO3和5 g无水乙醇作溶剂,回流反应12 h,减压浓缩,得到6.50 g棕黄色粘稠液体产物Ⅵ.FT-IR( ,cm1):3287(s),3078(s),2954(w),2928(s),2857(s),1745(s),1645(s),1 560(s),1 462(s),1 375(w),1 267(s),1 119(s),1 044(w),763(w),735(w).
2 结果与讨论
2.1 2-噁唑烷酮(Ⅰ)合成研究
2-噁唑烷酮(Ⅰ)的合成途径很多,如可由乙醇胺与碳酸乙烯酯[7-8]、碳酸二乙酯[9]、尿素[10]在不同的催化剂作用下反应制备.本文使用已活化的醋酸锌为催化剂,催化碳酸二甲酯(DMC)和乙醇胺反应生成2-噁唑烷酮,在这个反应中,胺基乙醇的胺基先进攻DMC分子中的羰基,形成线性胺基甲酸酯,进一步关环形成预期产物,该工艺后处理简单,如方程式1所示.
2.2 2-噁唑烷酮反应性能的研究
2-噁唑烷酮是一个多官能团化合物,如胺基上活泼氢可发生氮杂-麦克尔加成及羰基上的两种开环反应.Yang等[6]报道了2-噁唑烷酮在KF/Al2O3催化下与丙烯酸酯的反应,反应具有很强的溶剂效应.本文在80℃及K2CO3催化下本体反应2.5 h,几乎定量反应生成Ⅱ(1H NMR见图1).

方程式1 化合物Ⅰ的合成途径Equ.1 Pathway for thesynthesisof compoundⅠ
Ⅱ和1,6-己二胺发生酯的酰胺化反应生成两端为2-噁唑烷酮的化合物Ⅲ(方程式3),1HNMR见图2.根据核磁图中峰面积计算可以知道,还有约10%的原料没有反应.考虑到化合物Ⅲ的官能度为2,将其和另一个官能度为2的醇、胺反应,可以合成非异氰酸酯聚氨酯或聚脲.为此将Ⅲ继续和1,6-己二胺进行反应,最终得到黄色蓬松状、略有弹性的固体,反应过程中有气体放出.遗憾的是该产物可能发生了交联,不溶于水、二氯甲烷、氯仿、DMSO、乙醇、甲醇、甲苯、四氢呋喃、丙酮和浓硫酸等常用溶剂,无法对其进行核磁表征.

图1 化合物Ⅱ的1H NMR Fig.1 1H NMRspectrum ofⅡ

方程式3 化合物Ⅲ的合成Equ.3 Synthesisof compoundⅢ
为了进一步搞清反应的途径,由2-噁唑烷酮衍生物Ⅱ与丁胺反应合成单2-噁唑烷酮的酰胺产物Ⅴ(方程式4,1H NMR见图3).化合物Ⅴ为单官能团化合物,与1,6-己二胺反应从理论上讲不会交联,所得产物VI的1HNMR见图3.从图3两个化合物氢谱的对比中可以发现,归属于噁唑烷酮的信号a大大减小,反应后出现了3组新峰,分别为1峰、2峰和3峰.由于产物较复杂,分离困难.利用核磁软件模拟化合物Ⅵa、Ⅵc及Ⅵd的1HNMR发现,化合物Ⅵc特征峰的氢谱位置大部分集中在2.0~3.0ppm之间,而化合物Ⅵa和化合物Ⅵd特征峰的氢谱位置主要在3.0~4.0 ppm之间.产物核磁图中2峰和3峰可能归属予化合物Ⅵc,由峰面积可以知道化合物Ⅵc为主要产物.13C NMR(图4)中的3峰和4峰是Ⅵa分子上与羟基相连的两个碳原子(-CH2CH2OH)的信号峰,1峰是Ⅵd五元环中羰基碳原子(-NCON-)的信号峰,2峰是Ⅵb中羧基和胺形成盐的碳原子(-NCOO N+-)[11]的信号峰.由此可以推断,噁唑烷酮在该反应条件下开环反应选择性较差,可以形成带羟乙基的脲,环状脲基多胺化合物.可能的反应途径见(图5).

图2 化合物Ⅲ的1H NMRFig.2 1H NMRspectrum ofⅢ

方程式4 化合物Ⅴ的合成Equ.4 Synthesisof compoundⅤ

图3 化合物V和VI的1H NMRFig.3 1H NMRspectraof V and VI

图4 化合物Ⅵ的13CNMRFig.4 13CNMRspectrum ofⅥ

图5 胺与噁唑烷酮的可能反应途径Fig.5 Thepossiblepathway of aminereacted with oxazolidinone
为了考察交联产物Ⅳ与Ⅵ结构是否相似,两种Ⅳ及Ⅵ的红外光谱图见图6.
由图6可以看出,两种化合物在3000cm1以上都具有较强的N-H氢键信号,化合物Ⅳ中氢键相互作用比Ⅵ强,峰较宽,化合物Ⅵ中在1 745.96 cm1有较强的羰基信号,表明环状脲的形成[12],其它峰基本相似,表明化合物Ⅳ可能是聚合的多胺,环状的脲基较少.
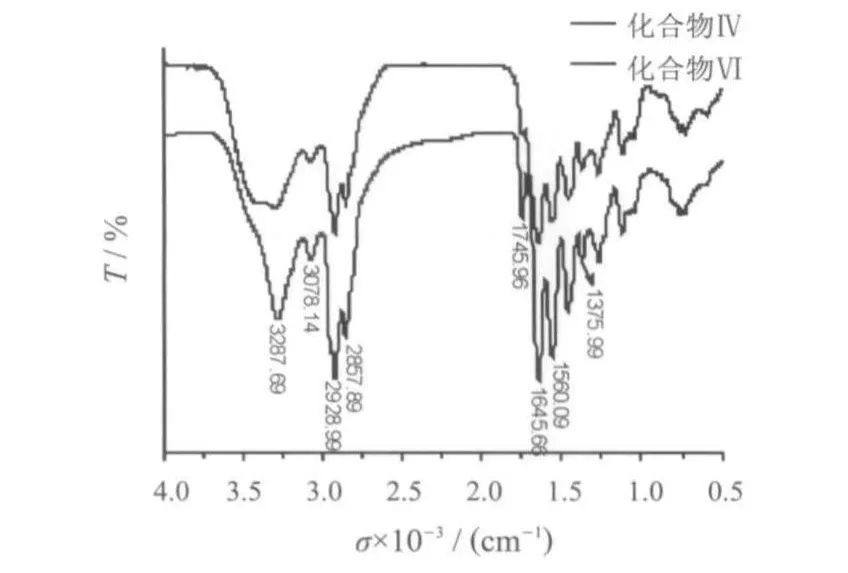
图6 化合物Ⅳ和Ⅵ的IR谱图Fig.6 FT-IRspectraof compoundⅣandⅥ
3 结论
在醋酸锌催化下乙醇胺和碳酸二甲酯反应生成噁唑烷酮,噁唑烷酮与丙烯酸甲酯发生麦克加成反应,形成3-噁唑烷酮丙酸甲酯,进一步与丁胺、1,6-己二胺发生酰胺化反应合成出了2-噁唑烷酮衍生物.通过研究发现,有机胺与环状氨基甲酸酯的羰基反应形成羟乙基取代脲,与烷氧基反应形成开环的胺基甲酸,该化合物进一步脱水形成环状脲,脱二氧化碳形成多胺化合物.
[1]Adams P,Baron FA.Estersof Carbamic Acid[J].Chem Rev,1965,65(5):567-602.
[2]Madesclaire M,Coudert P,Zaitsev V P,et al.Synthesisof 2-oxazolidinonesfrom (1S,2S)-2-amino-1-(4-nitrophenyl)-1,3-propanediol[J].Chem Heterocycl Compd(Engl Transl),2006,42(4): 506-511.
[3]李立清,王晓刚,郭三霞,等.一锅法有效合成氨基甲酸酯类化合物 [J].有机化学,2007,27(4):519-523.
[4]梅付名,梅慧,李光兴.羰基化法合成氨基甲酸酯催化剂的研究进展 [J].现代化工,2006,26(8):14-18.
[5]Rajca A,Grobelny D,Witek S,et al.5-Aryl-2-oxo-1,2,4-oxathiazolesas Cyclocarbonylating Agentsfor 2-Aminoalcoholsand 1,2-Diamines[J].Synthesis,1983,12:1032-1033.
[6]Yang L,Xu L-W,Xia CG.Highly efficient KF/Al2O3-catalyzed versatilehetero-Michael addition of nitrogen,oxygen,and sulfur nucleophilesto,-ethylenic compounds[J].Tetrahedron Lett,2005,46(19):3279-3282.
[7]Patil Y P,Tambade PJ,Jagtap SR,et al.Synthesis of 2-oxazolidinones/2-imidazolidinones from CO2,different epoxides and amino alcohols/alkylenediaminesusing Br Ph3+P-PE-P+Ph3Br ashomogenousrecyclablecatalyst[J].JMol Catal A: Chem,2008,289:14-21.
[8]Jagtap SR,Patil Y P,Fujita S-I,et al.Heterogeneous base catalyzed synthesis of 2-oxazolidinones/2-imidiazolidinones via transesteri?cation of ethylenecarbonatewith b-aminoalcohols/1,2-diamines[J].Appl Catal,A:Gen,2008,341:133-138.
[9]Gall EL,Hurvois J-P,Sinbandhit S.Regio-and diastereoselective synthesis of-cyanoamines by anodic oxidation of 6-membered -silylamines[J].Eur JOrg Chem,1999,1999(10):2645-2653.
[10]Bratulescu G.An excellent procedurefor thesynthesisof oxazolidin-2-ones[J].Synthesis,2007,20:3111-3112.
[11]Hampe EM,Rudkevich D M.Exploring reversiblereactionsbetween CO2and amines[J].Tetrahedron,2003,59(48):9619-9625.
[12]薛燕,陆世维.硒催化的邻硝基苯胺类化合物的羰基化反应 [J].化学通报,2002,3:187-190.
猜你喜欢
现代实用医学(2022年10期)2022-12-08
阅读(科学探秘)(2021年5期)2021-08-04
中国民间疗法(2021年9期)2021-07-22
发明与创新(2018年2期)2018-05-25
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
化工进展(2015年3期)2015-11-11
环球时报(2014-03-07)2014-03-07
组合机床与自动化加工技术(2014年12期)2014-03-01
无机化学学报(2014年5期)2014-02-28
无机化学学报(2014年5期)2014-02-28
