
间三氟甲基-N-羟乙基苯胺的合成研究
2011-08-24 00:56李晓芳李永龙唐守万潘富友
浙江工业大学学报 2011年6期
李晓芳,李永龙,唐守万,潘富友
(1.浙江工业大学 化学工程与材料学院,浙江 杭州 310032;2.台州学院 医药化工学院,浙江 台州 317000)
间三氟甲基-N-羟乙基苯胺的合成研究
李晓芳1,李永龙1,唐守万2,潘富友2
(1.浙江工业大学 化学工程与材料学院,浙江 杭州 310032;2.台州学院 医药化工学院,浙江 台州 317000)
以间三氟甲基苯胺(Ⅰ)作为原料,通过与氯甲酸-2-氯乙酯进行选择性的单酰胺化反应(Ⅱ)后,再在氢氧化钠水溶液中经水解重排反应,合成了间三氟甲基-N-羟乙基苯胺(Ⅲ),产物结构经1H NMR,MS和IR表征.考察了反应溶剂、NaOH用量、反应温度和反应时间对产物收率的影响,实验结果得到:酰胺化反应以二氧六环为溶剂,反应时间3 h,反应温度70℃;第二步(Ⅱ)水解重排反应的适宜反应条件为n(间三氟甲基苯胺)∶n(NaOH)=1∶6,反应温度为70℃,反应时间为5 h,Ⅲ的总收率可达61%.
间三氟甲基苯胺;氯甲酸-2-氯乙酯;间三氟甲基-N-羟乙基苯胺;合成
N-羟乙基苯胺及其衍生物(羟基经氨解、酯化、乙酰化、氯化、脱水缩合等反应制得)作为精细化学品中一类重要的有机合成中间体,一直得到人们的高度重视和深入研究,在高分子染料、有机颜料、染发剂、彩色液晶显示以及彩色摄影胶卷等的制备方面有着重要作用,如分散红 DR19(4-(N,N-二羟乙基)胺基-4′-硝基偶氮苯)就是一种经典的偶氮发色基团,其氮原子上的羟乙基可大大改进染料在合成纤维中的分散性能;另外,它还具有光致变色、液晶现象及非线性光学等特性,可作为光电子材料中的信息储存材料[1].目前,羟乙基苯胺的合成工艺路线主要有两种:氯乙醇法[2-5]和环氧乙烷法[4-6],但通过这两种方法制备的产物都为N,N-(二羟乙基)苯胺或N-取代-N-羟乙基苯胺,无法得到单N-羟乙基苯胺类化合物.迄今为止,对于单N-羟乙基苯胺类化合物,人们还没有研究出一种合适的高效的合成方法.2003年,Shi等[7]曾报道在高压条件下利用路易斯酸Sn(O3SCF3)2作为催化剂,利用间三氟甲基苯胺与环氧乙烷反应可同时制得间三氟甲基-N,N-(二羟乙基)苯胺(51%)和间三氟甲基-N-羟乙基苯胺(38%),但此法并没有得到广泛应用,原因在于其一是由于单N-羟乙基苯胺选择性较低且得到的产物难分离,二是该反应条件苛刻,需在高压下进行,而且环氧乙烷易燃、易爆.由此可见,对于合成单N-羟乙基苯胺,人们面临的首要问题是如何在较温和的条件下提高其选择性.
该实验设计以间三氟甲基苯胺Ⅰ和氯甲酸-2-氯乙酯为原料,分两步合成间三氟甲基-N-羟乙基苯胺Ⅲ.首先,Ⅰ与氯甲酸-2-氯乙酯反应生成中间体Ⅱ,该反应以1,4-二氧六环作为反应溶剂,加入CaCO3作为缚酸剂;Ⅱ滴加入NaOH水溶液中经水解重排得到产物Ⅲ,其单N-羟乙基化的选择性与文献[6]相比有较大提高,总收率61.0%,其结构经1H NMR,IR和MS表征,并对反应条件进行了优化.
1 实验部分
1.1 仪器与试剂
BUCHI M-565熔点仪;岛津 LC-10AT-VP高效液相色谱仪和FTIR-8400型傅里叶变换红外分光光度计(KBr压片);Quattro Micro API型液质联用仪(LC-MS);DF-101S集热式恒温加热磁力搅拌器,GS12-2型电子恒速搅拌仪.
间三氟甲基苯胺和氯甲酸-2-氯乙酯为工业品,纯度97%,其余所用试剂均为化学纯.
1.2 间三氟甲基-N-羟乙基苯胺的合成
间三氟甲基-N-羟乙基苯胺的合成路线,如图1所示.在250 mL干燥三口烧瓶中,加入35 mL无水1,4-二氧六环,16.1 g(0.1 mol)间三氟甲基苯胺,11.0 g(0.11 mol)碳酸钙,用电子恒速搅拌仪搅拌.冷至20℃,在搅拌状态下缓慢滴入15 g(1.05 mol)氯甲酸-2-氯乙酯,控制反应温度在50℃以下,滴加完毕升温至70℃,继续搅拌反应3 h.毕,将反应液倒入160 mL水中,抽滤除去白色粉末状固体,溶液分层得到的棕黄色油层缓慢滴入氢氧化钠水溶液中(24 g NaOH溶于60 mL水中),70℃下搅拌反应5 h.反应结束后,加入30 mL甲苯,用1∶1盐酸调节pH=10,油层分别用pH=9的碳酸钠溶液和盐水洗三次至中性,减压蒸馏得到淡黄色粘稠液体,静置后凝固为淡黄色针状物Ⅲ12.5 g,收率约61%,纯度97.6%(HPLC测定),m.p.40.4 ℃;1H NMR(200 MHz,CDCl3)δ:7.26(t,1 H,CH),7.00(d,1 H,CH),6.82(s,1H,CH),6.76(d,1 H,CH),4.23(s,1H,OH),3.87(d,2H,CH2),3.32(t,2H,CH2),1.85(s,1H,OH);IRυ(cm-1):3 398(O—H),3 030,3 091(Ph-H),2 945,2 860(CH2);MS m/z (%):206(M+,100).
1.3 色谱条件
该实验利用高效液相色谱确定反应终点和检测产物纯度.色谱柱:Sun Fire C18柱(5μm,250 mm×4.6 mm i.d.);流动相:V(甲醇)∶V(水)=80∶20;检测波长:230 nm;流速:1.0 mL/min;进样量:20μL.
2 结果与讨论
2.1 合成方法
文献[2-6]采用的环氧乙烷法和氯乙醇法均为合成N,N-双取代苯胺类化合物.Shi等[6]利用路易斯酸Sn(O3SCF3)2作为催化剂,用环氧乙烷法可制得间三氟甲基-N-羟乙基苯胺,但其收率较低,选择性较差,仅为38%.综合文献方法,该实验采用间三氟甲基苯胺和氯甲酸-2-氯乙酯作为原料合成中间产物Ⅱ,Ⅱ在NaOH水溶液中经水解重排反应得到产物间三氟甲基-N-羟乙基苯胺Ⅲ.与合成N,N-二羟乙基苯胺类化合物的环氧乙烷法和氯乙醇法比较,其不同之处在于氯甲酸-2-氯乙酯只取代其氮原子上的一个氢原子,得到单N-羟乙基苯胺类化合物,其选择性大大提高,这种方法提供了合成单N-羟乙基苯胺类化合物的有效途径.
2.2 缚酸剂的选择
间三氟甲基苯胺Ⅰ首先与氯甲酸-2-氯乙酯反应生成中间体Ⅱ的反应属于亲核取代反应,反应过程中产生的大量的酸,需要质子吸收剂,可用三乙胺、碳酸钾、碳酸钠及碳酸钙等作为缚酸剂,但不能用氢氧化钠作为缚酸剂,因为氢氧化钠在缚酸过程中产生水,会分解氯甲酸-2-氯乙酯,影响产率,三乙胺、碳酸钾及碳酸钠等在水中溶解,对后处理带来不便,CaCO3在缚酸后生成碳酸氢钙难溶于水,直接过滤可除去,因此,该实验选择加CaCO3作为缚酸剂.
2.3 反应溶剂的选择
间三氟甲基苯胺Ⅰ首先与氯甲酸-2-氯乙酯反应生成中间体Ⅱ,Ⅱ在NaOH水溶液条件下进行重排得到最终产物Ⅲ.第一步反应中溶剂的选择对合成中间体Ⅱ有很大影响,而Ⅱ的收率又会直接影响Ⅲ的收率,故全文分别比较了以1,4-二氧六环,四氢呋喃(THF)和吡啶为反应溶剂时对Ⅲ的收率的影响,结果列于表1.
从表1中可以看出,当选择THF和1,4-二氧六环作为反应溶剂,其他反应条件均相同时,Ⅲ的收率相对较高;而当选择吡啶作为溶剂时,即使第一步在回流温度下搅拌反应24 h,还有大量的苯胺未反应,导致Ⅲ的收率很低.由于二氧六环毒性比四氢呋喃和吡啶稍低,且当使用二氧六环时Ⅲ的收率最高,故该实验选择1,4-二氧六环作为第一步的反应溶剂,反应温度为70℃,反应时间为3 h.
2.4 水解重排反应条件的选择
该实验分别考察了NaOH用量、反应温度和反应时间对间三氟甲基-N-羟乙基苯胺Ⅲ收率的影响,确定合成间三氟甲基-N-羟乙基苯胺的适宜条件,结果如表2所示.
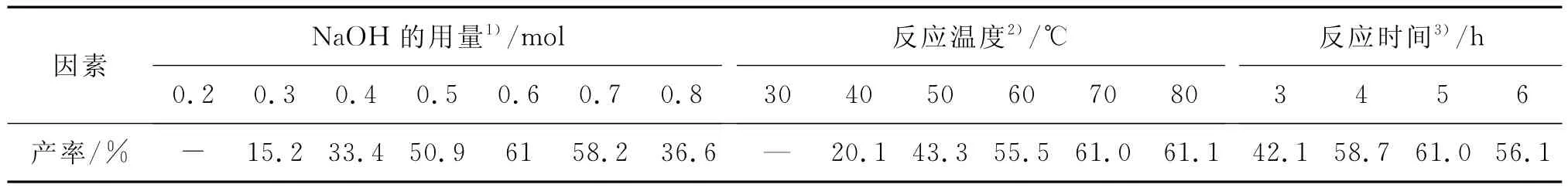
表2 影响重排反应的因素Table 2 Influence factors of rearrangement reaction
由表2可知,NaOH用量、反应温度和反应时间分别对Ⅲ的收率有较大影响.当反应温度为70℃,反应时间为5 h时,考察NaOH用量对收率的影响.NaOH用量为0.2 mol时,液相色谱动态跟踪检测结果显示无反应产物生成;随着NaOH用量的增加,间三氟甲基-N-羟乙基苯胺的收率明显提高,当NaOH用量为0.6 mol(即间三氟甲基苯胺与NaOH的摩尔比为1∶6),收率为61.0%,且质量较好;但当NaOH的量继续增加至0.7,0.8 mol时,Ⅲ的收率没有提高反而有所下降,分别下降为58.2%和36.6%.
另外,当NaOH用量为0.6 mol,反应时间为5 h时,适当提高反应温度,Ⅲ的收率也相应提高:当温度为30℃,水解重排反应基本不进行,液相色谱动态跟踪检测结果显示无反应产物生成;升温至70℃时,液相色谱动态跟踪检测发现反应较好,且无副反应发生;当温度进一步提高升至80℃,其收率并没有明显增加.故确定适宜的水解重排反应条件为间三氟甲基苯胺与NaOH的摩尔比为1∶6,反应温度为70℃.
在上述适宜条件下(NaOH 0.6 mol,温度70℃),进一步考察了反应时间对反应收率的影响.反应进行4 h时,Ⅲ的收率为58.7%;反应5 h时,液相色谱跟踪检测不到中间体Ⅱ对应峰,说明Ⅱ已反应完全,此时,Ⅲ收率为61%;当反应时间进一步延长到6 h,收率反而下降,这可能是因为随着反应时间的延长,逐渐有聚合物生成.
综合上述结果,第二步水解重排反应的适宜反应条件:间三氟甲基苯胺与NaOH的摩尔比为1∶6,反应温度为70℃,反应时间为5 h.
3 结 论
按图1所示合成路线,采用两步法合成了间三氟甲基-N-羟乙基苯胺.研究表明,此反应较为适宜的反应条件:以1,4-二氧六环为溶剂,n(间三氟甲基苯胺)∶n(氯甲酸-2-氯乙酯)=1∶1.05,添加CaCO3作为缚酸剂,于70℃反应3 h合成Ⅱ;Ⅱ的水解重排反应适宜反应条件为n(间三氟甲基苯胺)∶n(NaOH)=1∶6,反应温度为70℃,反应时间为5 h.得到Ⅲ的总收率为61.0%.该合成方法可成功的仅将氮原子上的一个氢原子取代为羟乙基,这也可广泛应用于其他单N-羟乙基苯胺类化合物的合成.
[1]贾叙东,杨庆,余学海.用扩链法合成侧链含偶氮苯的液晶聚氨醇及表征[C]//中国化学会高分子委员会.全国高分子学术论报告会论文集.合肥:中国科学技术大学出版社,1997:54-55.
[2]余小五.新型聚二乙炔非线性光学材料合成[D].合肥:安徽大学高分子化学与物理系,2004.
[3]姚海波,王木立,章于川,等.一类芳香族偶氮化合物的合成及表征[J].合成化学,2003,11:156-159.
[4]程芳琴,杨凤玲,沈桂芝.N-苄基-N-甲基乙醇胺[J].精细与专用化学品,2003,7:24-25.
[5]杨凤玲,师红娟.N-乙基-N-羟乙基苯胺的合成及应用[J].石家庄化工,1999,4:1-3.
[6]申凯华,张蓉,刘建民,等.偶氮型聚氧乙烯醚分散染料结构的电喷雾质谱分析[J].分析化学,2001(1):32-34.
[7]SHI Min,CHEN Yu.Lewis acids catalyzed ring-opening reactions of methylenecyclopropanes and epoxides in supercritical carbon dioxide or modified supercritical carbon dioxide with perfluorocarbon[J].Journal of Fluorine Chemistry,2003,122(2):219-227.
Synthesis of 3-trifluoromethyl-N-hydroxyethylaniline
LI Xiao-fang1,LI Yong-long1,TANG Shou-wan2,PAN Fu-you2
(1.College of Chemical Engineering and Materials Science,Zhejiang University of Technology,Hangzhou 310032,China;2.College of Pharmaceutical and Chemical Engineering,Taizhou University,Taizhou 317000,China)
3-trifluoromethyl-N-hydroxyethylaniline was synthesized by esterification of 3-trifluoromethylaniline with chloroethyl chloroformate,then hydrolyzed and rearranged by treatment with NaOH.The structure of product was conformed by1H NMR,IR,and MS.The influences of reaction solvent,amount of NaOH,reaction temperature,and reaction time on the yield were investigated.The yield ofⅢcan reach 61%under the optimal reactions conditions:the reaction ofⅠwas kept at 70℃for 3 h in dioxane at first step,and n(Ⅰ)∶n(NaOH)with 1∶6 at 70℃for 5 h at second step for hydrolyzed and rearrangement.It provides an effective method for synthesizing monosubstitution-N-hydroxyethylaniline.
3-trifluoromethylaniline; 2-chloroethyl chloroformate; 3-trifluoromethyl-N-hydroxyethylaniline;synthesis
O625.63
A
1006-4303(2011)06-0596-04
2010-06-28
国家自然科学基金资助项目(20905057)
李晓芳(1986—),女,浙江缙云人,硕士研究生,主要从事精细有机合成的研究.通信作者:潘富友教授,E-mail:panfy@tzc.edu.cn.
(
陈石平)
猜你喜欢
能源化工(2022年1期)2023-01-14
中老年保健(2022年3期)2022-11-21
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
石油化工自动化(2020年1期)2020-03-05
枣庄学院学报(2019年5期)2019-09-20
通信技术(2019年8期)2019-09-03
中国循证儿科杂志(2019年2期)2019-06-04
分析化学(2017年12期)2017-12-25
中国生化药物杂志(2015年4期)2015-07-07
