
利用MLST技术对浙江省大肠杆菌O157的分子流行病学研究*
2011-08-21 10:23:38叶菊莲梅玲玲姚苹苹姜理平
中国人兽共患病学报 2011年10期
叶菊莲,占 利,梅玲玲,罗 芸,姚苹苹,姜理平
利用MLST技术对浙江省大肠杆菌O157的分子流行病学研究*
叶菊莲,占 利,梅玲玲,罗 芸,姚苹苹,姜理平
目的 对浙江省2005-2010年大肠杆菌O157分离株进行分子分型研究,了解菌株间的遗传进化关系,为浙江省大肠杆菌O157监测及爆发疫情的控制提供基础。方法 选择Pasteur大肠杆菌多位点序列分型(Multilocus Sequence Typing,MLST)方案,对大肠杆菌O157分离株及882364菌株进行 MLST分型,确定菌株序列型(Sequence type,ST);采用DNAsp、eBURST、START2等软件进行分析。结果 30株菌中,27株O157∶H7菌株具备相同序列型(ST-284),占90%(27/30),其他3株菌序列型分别为 ST-367、ST-125、ST-296。8个管家基因核苷酸多态性(Pi)范围为0.00119(polB)~0.00648(uidA),putP基因多态性位点比例最高(4.6%),trpA基因多态性位点比例最低(0.4%)。进化分析结果显示,这些序列型分别属于Group 1及Group 11群。结论 浙江省动物源性大肠杆菌O157∶H7的主要序列型为ST-284,与江苏省病人株882364(ST-296)具有同一个进化祖先,提示应加强浙江省大肠杆菌O157病原学监测。
大肠杆菌O157;多位点序列分型(MLST);管家基因;分子流行病学
自大肠埃希菌O157被认识以来,对其基因的研究越来越细,目前已探明了许多结构与功能基因,并发现存在许多变异株,在其病因学、病理学及临床治疗方面均有长足的进展。随着细菌食源性疾病爆发的日益频繁以及多重耐药菌株的不断涌现,细菌形态、血清型和噬菌体型等传统分型方法已不能满足疫情分析及流行病学研究需要。常用的PFGE一般用于传染病爆发调查,而无法对菌株的遗传进化特征进行分析;MLST法则适宜于菌株的进化关系及微生物群体遗传学分析,一般用于广义的流行病学研究[1-2]。为了解浙江省分离的大肠埃希菌O157菌株的进化关系及其群体遗传学特征,本研究选择大肠杆菌dinB、icdA、pabB、polB、putP、trpA、trpB、uidA 8个管家基因进行多位点序列分型(MLST),并对分型结果进行分析,结果报告如下。
1 材料和方法
1.1 实验菌株 29株菌均为浙江省疾病预防控制中心2005-2010年从各种动物及病人粪便标本中分离保存的O157菌株,882364为江苏省分离的O157∶H7菌株。详细信息见表1。
1.2 主要试剂 API20E生化鉴定条(法国梅里埃公司),大肠埃希菌诊断血清(日本生研株式会社)。PrimeSTARTMHS DNA Polymerase(2.5U/μL)聚合酶、dNTPs、100bp DNA Marker(大连宝生物工程有限公司),琼脂糖(西班牙Biowest),所有试剂均在有效期内使用。
1.3 仪器 Master cycler gradient PCR扩增仪;FR-200A全自动紫外与可见分析装置(上海复日科技有限公司);Minirun GE-100电泳仪(杭州博日科技有限公司),热裂解仪(杭州博日科技有限公司,HB-100)。
1.4 生化鉴定分型 所有菌株均用API20E生化条进行系统鉴定,符合大肠菌群特性者进行血清分型。
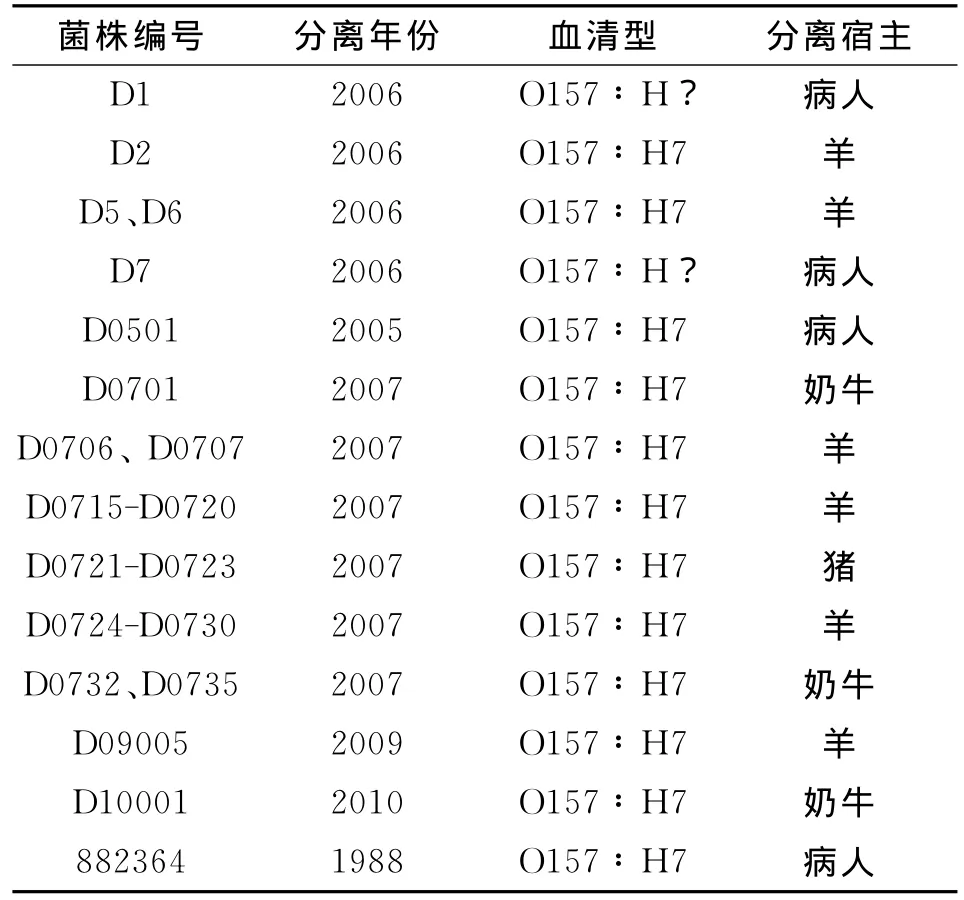
表1 30株大肠杆菌O157菌株信息Table 1 Origins,serotypes and hosts of E.coli O157isolates
1.5 引物合成 参考文献[3-4]进行引物合成,所有引物均由上海生工生物工程技术服务有限公司合成。
1.6 模板制备 取平板新鲜分离纯菌,悬浮于1mL无菌纯水中,置热裂解仪,100℃,20min,12 000r/min离心5min,取上清液作为DNA模板,-20℃冰箱备用。
1.7 PCR扩增靶基因 50μL反应体系包括0.5 μL PrimeSTARTMHS DNA 酶,上下游引物(10μmol/L)各1μL ,模板5μL。PCR扩增条件参考[3]。取PCR扩增产物3μL以琼脂糖凝胶电泳进行检测。
1.8 DNA测序及MLST数据分析 取50μL PCR扩增产物送北京六合华大基因科技股份有限公司进行测序,测序结果使用Chromas及DNA Star软件进行修正后,提交Pasteur在线数据库进行处理,获得各管家基因的等位基因编码(allele number),并形成各菌株的序列型。DNAsp5.0分析各管家基因的多态性,eBURST、START2等软件分析菌株间的进化关系。
2 结 果
2.1生化反应 用法国梅里埃肠杆菌科API 20E生化反应条鉴定,根据反应编码查对,所有菌株均为大肠埃希菌,血清分型结果详见表1。
2.2 PCR结果 经条件优化后,所有受试菌株均扩增出各管家基因并进行测序分析。
2.3 核苷酸多态性分析 使用DNAsp 5.0软件分析本研究中30株大肠杆菌8个管家基因的核苷酸多态性(以Pi表示)。结果见表2。
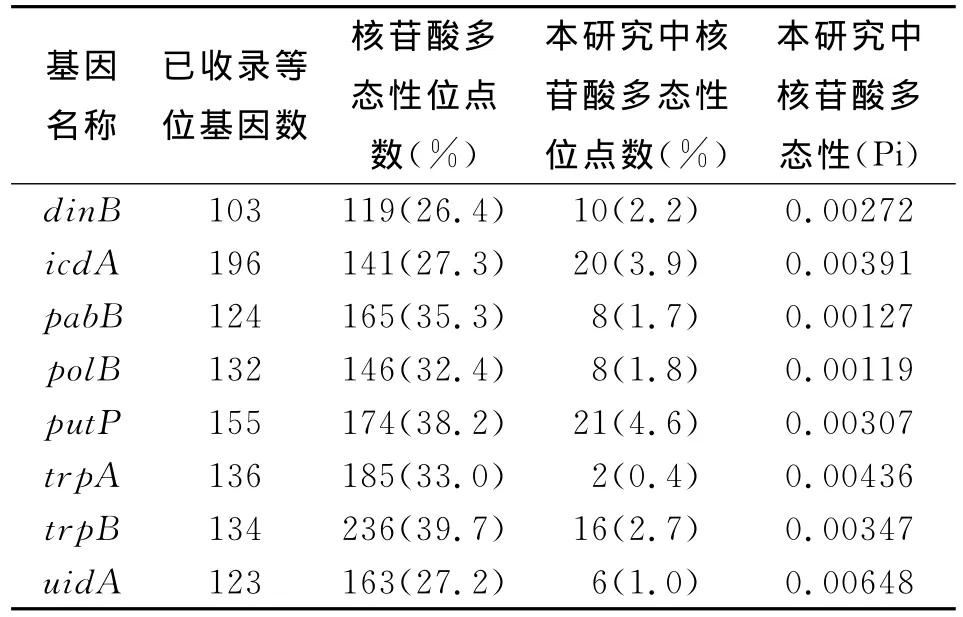
表2 30株大肠杆菌O157 8个管家基因核苷酸多态性分析结果Table 2 Nucleotide polymorphism found in the eight housekeeping genes used for MLST among 30 E.coli O157 isolates
由此可见,8个管家基因在已收录的数据库中,trpB存在的多态性位点比例最高(39.7%),dinB存在的多态性位点比例最低(26.4%);而本次所研究的30株EHEC O157中,putP存在的多态性位点比例最高(4.6%),trpA存在的多态性位点比例最低(0.4%),这些基因的核苷酸多态性(Pi)范围为0.00119(polB)~0.00648(uidA)。
2.4 MLST分型结果 根据Chromas图谱及各等位基因长度,对所得序列进行修正,置于Pasteur MLST数据库中查询各管家基因的等位基因数值,进而获得菌株的ST型。2005-2010年浙江省分离的29株菌株中27株为相同序列型ST-284(59-110-88-63-93-84-83-86),D1、D7及江苏省的882364的序列型 分 别为 ST-367(8-104-7-3-7-1-2-4)、ST-125(10-2-4-3-18-1-23-4)、ST-296(68-110-91-63-93-84-83-86);其中ST-284序列型所占比例最多,占90%(27/30)。
2.5 数据分析 设定群所需最小相同等位基因数(Minimum No.of identical loci for group definition)为5,取样自展值(No.re-samplings for bootstrapping)设置为1000,获得以下两组eBURST分析结果(见图1、2)。可见ST-367和ST-125同属于Group 1 群,而 ST-284 和 ST-296则属于 Group 11群。
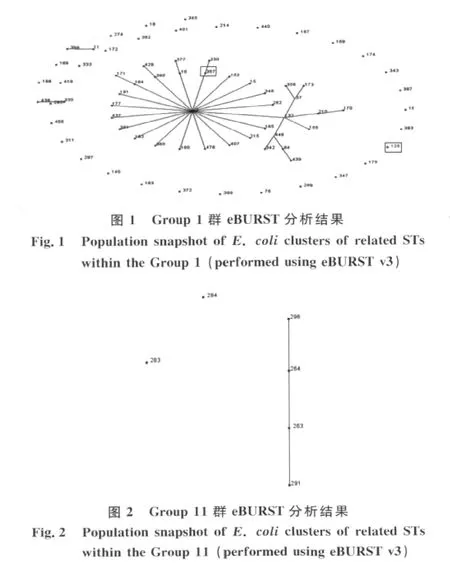
Jaureguy等研究结果显示,ST-192,ST-246,ST-263,ST-264为Pasteur数据库中出血性大肠杆菌的序列型,其中ST246为O26的EHEC的ST型,ST157为一株ETEC的序列型。使用START V2.0软件,选择UPGMA对这些进行遗传进化分析。结果显示 ST-284、ST-263、ST-264、ST-296位于进化距离比较近的同一分支上,而ST125和ST367则位于另一距离较近的分支上(见图3)。
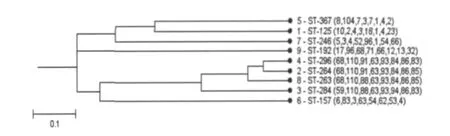
图3 30株出血性大肠杆菌O157(START V2.0)进化树Fig.3 MLST dendrogram of E.coli isolates(performed using START V2.0)
3 讨 论
近年来,由出血性大肠杆菌(enterohemorrhagic Escherichiacoli,EHEC)引起的食源性疾病在世界范围内许多国家都时有报道,如最近发生的日本烤肉店生牛肉中毒事件的罪魁祸首就是肠出血性大肠杆菌O111,以及今年5月份发生在德国由豆苗污染引起的肠出血性大肠杆菌O104∶H4暴发事件,到目前为止已导致2000多人感染,约数十人死亡,这次疫情对整个欧洲都造成了一定的影响。EHEC主要包括O157∶H7、O26∶H11和O111∶MN等血清型,O157∶H7是最重要的一种血清型[5-7]。该菌常见于牛、羊等温血动物的肠内。1998年,浙江省开始大肠杆菌O157监测,并先后在嘉兴、金华、衢州、杭州、舟山、绍兴等地市牛、羊、猪等多种动物及腹泻患者粪便中检出多株大肠杆菌O157菌株。由于浙江省在地理位置上比邻江苏省及安徽省,而江苏省及安徽省曾经出现疫情,为了有效的防止浙江省大肠杆菌O157疫情的发生,必须及时了解浙江省大肠杆菌O157病原体的分子流行病学状况,特别是菌株之间的遗传进化关系。多位点序列分型可分析不同时间不同地区临床分离株的遗传相关性,特别适用于多地域相关性微生物的流行病学、种内微进化、来源追踪和抗生素耐受等的研究。它克服了不同实验室数据重复性及可比性差的缺点,直接利用所测定的管家基因的核苷酸序列组合进行编码,技术流程标准化,结果易于验证,数据便于电脑保存,并可通过互联网共享[8,2]。与PFGE相比,更适宜于菌株的进化关系及微生物群体遗传学分析,可用于广义的流行病学研究[1,8]。
目前,适用于大肠杆菌MLST分析的数据库一共有3个[9],除本研究选用的Pasteur研究中心的数据库外,还有 Whittam数据库(http://www.shigatox.net/ecmlst/cgi-bin/dbquery)及 Achtman数据库(http://mlst.ucc.i.e/mlst/dbs/Ecoli)[5],这3个数据库用来分型的等位基因组合不尽相同。根据Gordon[10]的研究显示,这3个数据库的数据存在极高的关联性,说明大肠杆菌的克隆结构是十分健全的,这3个数据库之间的共享及相互之间的联系正在研究中。由于Pasteur研究中心的MLST方案依据的是8个管家基因序列,相比前2个数据库,纳入分型范围内的基因种类及长度更多,因此,本研究选择适用该方案进行浙江省大肠杆菌O157的遗传性研究。
Clermont等 研 究 显 示[4,9,11],不 同 血 清 型 的 大肠杆菌的8个管家基因核苷酸多态性位点及核苷酸多态性会存在一定差异,本研究结果与其相符。核苷酸多态性分析发现,浙江省所分离的出血性大肠杆菌O157菌株中,管家基因uidA具有最大的核苷酸多态性(Pi)(0.00648),而putP基因具有最高比例的多态性位点(4.6%),与Jaureguy等[4]的研究数据存在一定差异。这可能与本实验中的菌株均为出血性大肠杆菌O157以及地区差异有关。本研究中27株大肠杆菌O157∶H7具有相同序列型ST-284;菌株编号为D1、D7、882364的是不同的序列型,分 别 为 ST-367、ST-125、ST-296。 根 据eBURST和START 分析结果,ST-284、ST-296同属于Group 11,具有相同的进化祖先,而ST-367、ST-125同属于Group 1,处于同一进化分支上。这两个群在等位基因存在较多差异,其进化关系较远。从而判断大肠杆菌O157∶H7与O157:H?处于不同的进化分支上,即不同血清型的菌株起源于不同的祖先,但同一血清型的菌株一般起源于同一个祖先。同时发现分离自我省动物体内的大肠杆菌O157∶H7与江苏省O157∶H7病人源性菌株具有同一进化祖先,仅dinB、icdA存在差异,而根据DNAsp分析结果显示,这两个管家基因具有较高的多样性,因此建议继续加强浙江省大肠杆菌O157∶H7的病原学监测。而浙江省近年来散发的O157病人分离菌株属于同一群,具有相同的进化祖先,这也需要引起重视,注意监测大肠杆菌O157的遗传进化关系的变换。本研究采用MLST分型技术,初步分析了浙江省大肠杆菌O157的遗传进化关系,但由于标本量少,是否还有其他新的ST型还有待进一步研究。
[1]王中强,邱少富,王勇,等。多位点序列分型技术及其研究进展[J].军事医学院院刊,2010,34(1):76-79.
[2]谢蟪旭,王萍.多位点序列分型法在感染性疾病中的应用[J].国际口腔医学杂志,2009,36(5):557-560.
[3]Genotyping of Pathogens,Public Health Platform(PF8).Primers used for MLST ofEscherichiacoli[EB/OL].[2010-09-20].http://www.pasteur.fr/recherche/genopole/PF8/mlst/primers_Ecoli.html.
[4]Jaureguy F,Landraud L,Passet V,et al.Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains[J].BMC Genomics,2008,26(9):560-573.
[5]Riley LW,Remis RS.Helgerson SD,et al.Hemorrhagic colitis associated with a rareEscherichiacoliserotype[J].N Engl J Med,1983,308(12):681-685.
[6]Dunn JR,Keen JE,Moreland D,et al.Prevalence ofEscherichiacoliO157∶H7in white-tailed deer from louisiana[J].J Wildl Dis,2004,40(2),361-365.
[7]Fenq PC,Keys C,Lacher D,et al.Prevalence,characterizationand clonal analysis ofEscherichiacoliO157:non-H7serotypes that carryeae alleles[J].FEMS Microbiol Lett,2010,308(1)62-67.
[8]Maiden MC.Multilocus sequence typing of bacteria[J].Annu Rev Microbiol,2006.60:561-588.
[9]Clermont O,Olier M ,Hoede C,et al.Animal and human pathogenicEscherichiacolistrains share common genetic backgrounds[J].Infect Genet Evol,2011,11(3):654-662.
[10]Gordon DM,Clermont O,Tolley H,et al.AssigningEscherichia colistrains to phylogenetic groups:multilocus sequence typing versus the PCR triplex method[J].Environ Microbiol,2008,10(10):2484–2496.
[11]Ji XW,Liao YL,Zhu YF,et al.Multilocus Sequence Typing and Virulence Factors Analysis ofEscherichiacoliO157Strains in China[J].J Microbiol,2010,48(6):849-855.
Multilocus sequence typing analysis application in molecular epidemiological research ofEscherichiacoliO157isolates in Zhejiang province
YE Ju-lian,ZHAN Li,Mei Ling-ling,LUO Yun,YAO Ping-ping,JIANG Li-Ping
(ZhejiangCenterforDiseaseControlandPrevention,Hangzhou310051,China)
The objective was to investigate the molecular epidemiological feature ofEscherichiacoliO157isolates in Zhejiang Province.A multilocus sequence typing(MLST)scheme was applied to identify sequences of 8housekeeping genes including DNA polymerase(dinB),isocitrate dehydrogenase(icdA),p-aminobenzoate synthase(pabB),polymerase PolII(polB),proline permease,(putP),tryptophan synthase subunit A(trpA),tryptophan synthase subunit B(trpB)and beta-glucuronidase(uidA)from 29EHEC O157isolated between 2005and 2010in Zhejiang Province and 882364isolated in Jiangshu Province.DNAsp,eBURST and START2were applied to analyze the polymorphism of the eight housekeeping genes and the evolution of the EHEC O157.Four sequence types(STs),named ST-284,ST-367,ST-125,and ST-296were identified,which grouped into two lineages,Group 1and Group 11.ST-284had the most proportion of 90%(27/30).The proportion of variable sites ranged from 0.4%(trpA)to 4.6%(putP).The nucleotide diversity(Pi,average number of nucleotide differences per site between two randomly-selected isolates)ranged from 0.00119(polB)-0.00648(uidA).Phylogenetic analysis suggested that these STs belong to two CCs(clone complex).The data presented here provide new sights into molecular epidemiology feature of EHEC O157.MLST molecular typing of EHEC O157isolates in Zhejiang Province call for intense surveillance efforts.It was an ideal tool to the investigation of the genetic evolution of.EHEC O157.
enterohaemorrhagicEscherichiacoli(EHEC)O157;multilocus sequence typing(MLST);House-keeping gene;molecular epidemiology
R18
A
1002-2694(2011)10-0901-04
*国家科技重大专项:艾滋病和病毒性肝炎等重大传染病防治(2009ZX10004-210)
占利,Email:lzhan@cdc.zj.cn
浙江省疾病预防控制中心微生物所,杭州 310051
2011-06-21;
2011-08-10
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
基层中医药(2020年5期)2020-09-11 06:32:00
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
基层中医药(2018年5期)2018-08-31 02:35:42
西南农业学报(2016年6期)2016-04-16 05:12:47
法医学杂志(2015年4期)2016-01-06 12:36:36
制造技术与机床(2015年10期)2015-04-09 07:06:14
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
中国中医药现代远程教育(2014年21期)2014-03-01 04:32:23
