
肥大细胞在肥胖相关性肾病肾组织损伤中的作用
2011-08-07 08:16:16王旭方张明超郑春霞侯金花葛永纯谢红浪刘志红
肾脏病与透析肾移植杂志 2011年4期
王旭方 张明超 郑春霞 陈 浩 侯金花 葛永纯 谢红浪 刘志红
近年来,我国肥胖的发生率正逐年升高。作为心血管疾病、糖尿病、高血压、脂质代谢紊乱等疾病的重要危险因素,肥胖已成为威胁我国公共卫生的重大问题[1]。肥胖除了作为慢性肾脏病和终末期肾功能衰竭的危险因素外,还可以引起蛋白尿、肾组织损伤及肾功能减退,即肥胖相关性肾病(obesity related glomerulopathy,ORG)[2]。
我们以往的工作显示,我国ORG的发病率呈快速上升趋势[3]。足细胞密度和数量的下降与ORG蛋白尿及肾功能损伤呈显著相关性[4],控制体质量指数(body mass index,BMI)有助于蛋白尿的缓解。但同时我们也发现,并非所有BMI和尿蛋白下降患者其肾功能都维持稳定或得到一定程度恢复,部分肾活检时存在较重的肾小管萎缩及间质纤维化病变者,其肾功能预后不佳;进一步分析肾功能的影响因素,证实肾小管萎缩和间质纤维化对肾功能进展有预测作用[5]。然而目前ORG肾小管损伤机制尚不清楚,零星报道认为肾脏局部肾素-血管紧张素系统(renin-angiotensin system,RAS)激活、脂质沉积和氧化应激等可能对肾小管损伤有一定作用[6,7]。
随着研究深入,越来越多的证据表明炎症在ORG的发生和发展中发挥重要功能。ORG肾小球基因谱的研究发现有关炎性因子的基因表达明显上调,表明炎症反应在ORG的发生和发展中可能有重要作用[8]。肥胖患者脂肪组织巨噬细胞能够分泌肿瘤坏死因子 α(tumor necrosis factor-α,TNF-α)、白细胞介素 6(interleukin-6,IL-6)和 IL-1β等多种炎性因子[9],通过诱发胰岛素抵抗、促进细胞游离脂肪酸摄取及促进动脉粥样硬化形成等间接影响肾脏结构及功能[10]。体内实验证实,肥大细胞也参与了肥胖个体脂肪组织炎症反应[11]。肥大细胞敲除或使用肥大细胞稳定剂,可使小鼠脂肪组织巨噬细胞数量减少,炎性因子水平显著下降,体重减轻。但肥大细胞在ORG中发挥何种功能目前尚无研究报道。
基于以上研究,我们对ORG患者肥大细胞的数量、分布特点及其与临床病理特征的联系进行观察,探讨肥大细胞与ORG肾组织损伤及病情进展之间的关系。
对象与方法
病例选择 结合临床表现、实验室检查及肾活检确诊的ORG患者39例,其中男性23例,女性16例,均为中年患者。全部患者满足以下条件:(1)BMI≥28 kg/m2[3],排除内分泌性、药物性肥胖,排除糖尿病,可伴有空腹血糖升高(<7.0 mmol/L)或糖耐量异常;(2)临床表现为不同程度的蛋白尿(尿蛋白排泄量>0.4 g/24h),无肉眼血尿,镜下血尿<10万/ml;(3)肾脏病理表现为肾小球肥大,伴或不伴有局灶节段性肾小球硬化(FSGS),免疫荧光染色为寡免疫复合物沉积,可伴有IgM、C3非特异性或节段沉积;(4)结合临床、病理改变,除外肥胖患者合并有其他肾脏疾病,如IgA肾病、膜性肾病、糖尿病肾病、高血压肾损害等;(5)排除支气管哮喘、药物过敏及3个月内有感染的患者;同时选取10例性别年龄匹配的健康移植供肾组织标本作为正常对照,所选供者无高血压、肥胖、糖脂代谢紊乱、支气管哮喘及药物过敏等病史,组织病理分析无明显病变。
临床资料采集 详细采集病史,记录研究对象肾活检时性别、年龄、肾脏病病程、既往用药史,特别是血管紧张素转换酶抑制剂(ACEI)/血管紧张素Ⅱ受体拮抗剂(ARB)的使用情况。体格检查记录身高、体重、血压。常规方法测定24小时尿蛋白定量、尿N-乙酰-β-D葡萄糖苷酶(NAG)、尿视黄醇结合蛋白(retinol binding protein,RBP)、13h 禁水禁食尿渗量,采用日立7180自动生化分析仪检测空腹血糖、尿素氮、血清肌酐(SCr)、尿酸、总胆固醇、三酰甘油、高密度脂蛋白(HDL)、低密度脂蛋白(LDL)、C反应蛋白(CRP),记录IgE水平。
高血压定义为收缩压≥140 mmHg和(或)舒张压≥90 mmHg;空腹血糖受损为6.1 mmol/L≤空腹血糖<7.0 mmol/L,糖耐量异常为7.8 mmol/L≤餐后2h血糖<11.1 mmol/L两者均可称为糖代谢异常;血脂紊乱为总胆固醇>6.2 mmol/L,或三酰甘油>2.2 mmol/L,或 LDL>3.1 mmol/L,或 HDL<1.0 mmol/L;肾功能减退为采用MDRD公式计算的肾小球滤过率(eGFR)<90 ml/(min·1.73m2)及 SCr>109.6 μmol/L;尿蛋白定量>3.5 g/24h为大量蛋白尿。
肾活检组织病理 肾脏穿刺活检组织经甲醛固定,石蜡包埋,切片厚度为2 μm,常规行 HE、PAS、PASM-Masson和Masson三色染色。免疫病理采用冰冻切片直接免疫荧光法。
肾组织类胰蛋白酶(tryptase)及CD68免疫组织化学染色 tryptase存在于肥大细胞分泌颗粒中,是肥大细胞的特异性标志[12],因此本研究选取tryptase染色阳性细胞作为肥大细胞,同时以CD68染色阳性细胞为巨噬细胞进行观察。石蜡切片脱蜡,用3%H2O2封闭内源性过氧化物酶10 min,10 mmol/L柠檬酸盐缓冲剂(pH 6.0)条件下高温高压修复抗原,再加入10%小牛血清封闭。加入鼠抗人tryptase 抗体(1∶1 000,MAB-1222,Chemicon)或鼠抗人 CD68 抗体(1∶150,PG-M1,Dako),室温孵育 2h,PBS冲洗3遍,然后加二抗EnVision(Dako)孵育30 min。以 DBA显色,苏木素复染核,中性树胶(Sigma)封片,置显微镜(Olympus BX51)下观察。以同等浓度的非免疫兔血清IgG(Dako)替代一抗作为阴性对照。
肾组织tryptase及CD68双重免疫荧光染色石蜡切片如前步骤脱蜡、修复抗原后,加入Avidin/biotin各15 min,10%小牛血清封闭10 min,加鼠抗人 CD68(Dako,1∶150)室温孵育 2h,加生物素化抗鼠IgG孵育30 min,PBS漂洗三次,再加德克萨斯红Avidin DCS(Vector Laboratories,CA,1∶300)孵育 10 min,PBS冲洗 30 min。加鼠抗人 tryptase一抗(Chemicon,1∶1 000)孵育2h,加 FITC 标记的兔抗鼠IgG孵育30 min。甘油封片,共聚焦荧光显微镜(LSM510,Carl Zeiss)下观察并扫描采集图像。
结果判断 所需观察的病理切片由2名肾脏病理医师盲法评估,详细记录肾小球球性硬化、节段硬化、近端肾小管萎缩、间质纤维化、间质炎症细胞浸润、小动脉透明变性等情况。肾小球病变以病变小球占肾组织总小球数的百分比记录,肾小管间质病变根据皮质区病变累及的面积对每种病变进行半定量评分,受累面积<1%为0分,1~25%为1分,25% ~50%为2分,>50%为3分。细胞计数:在400倍放大显微镜下,以目镜测微器观察16个连续不重叠的肾皮质(包括肾小管间质及肾小球周围)视野,避开肾小球或大血管,所得数值之和为每mm2肥大细胞或巨噬细胞的个数。每张切片观察48个视野,取3次观察的平均值为最终结果。
统计分析 采用SPSS 13.0统计软件对数据资料进行统计分析,以单样本Kolmogorov-Smirnov Z检验对相关数据进行正态性检验,计量资料以均数±标准差或中位数表示,组间比较采用t检验;计数资料以百分率或构成比表示,组间比较采用χ2检验;变量间的相关性以pearson、spearman等相关系数评价;均为双侧检验,P<0.05为有统计学差异,P<0.01时差异非常显著。
结 果
临床特征 39例患者平均BMI(32.3±3.5)kg/m2。肾脏损害以蛋白尿为主,平均蛋白尿水平0.93 g/24h,其中3例患者表现为大量蛋白尿。平均eGFR(103.4±29.3)ml/min,其中2例患者eGFR<60 ml/(min·1.73m2)。11例患者肾活检病理表现为FSGS样病变,其余患者均表现为单纯肾小球肥大。ORG患者与对照组年龄及性别组成无明显差异,但29例合并高血压,22例合并糖代谢异常,34例合并血脂紊乱,3例患者血清IgE水平升高。
肥大细胞数量及分布特点 ORG患者肾组织肥大细胞浸润数量显著高于对照组[(16.81±10.99)vs(6.33 ±2.51),P<0.01](图 1),其主要分布于皮质区的肾间质,以萎缩的肾小管和间质纤维化处更多见,并可见肾小管炎,多见于萎缩的肾小管(图2),髓质区肥大细胞数量较少。肾小球内未见肥大细胞,但球囊周围可见肥大细胞浸润,部分小血管周围也可见肥大细胞浸润,肾间质可见较多脱颗粒的肥大细胞(图3)。
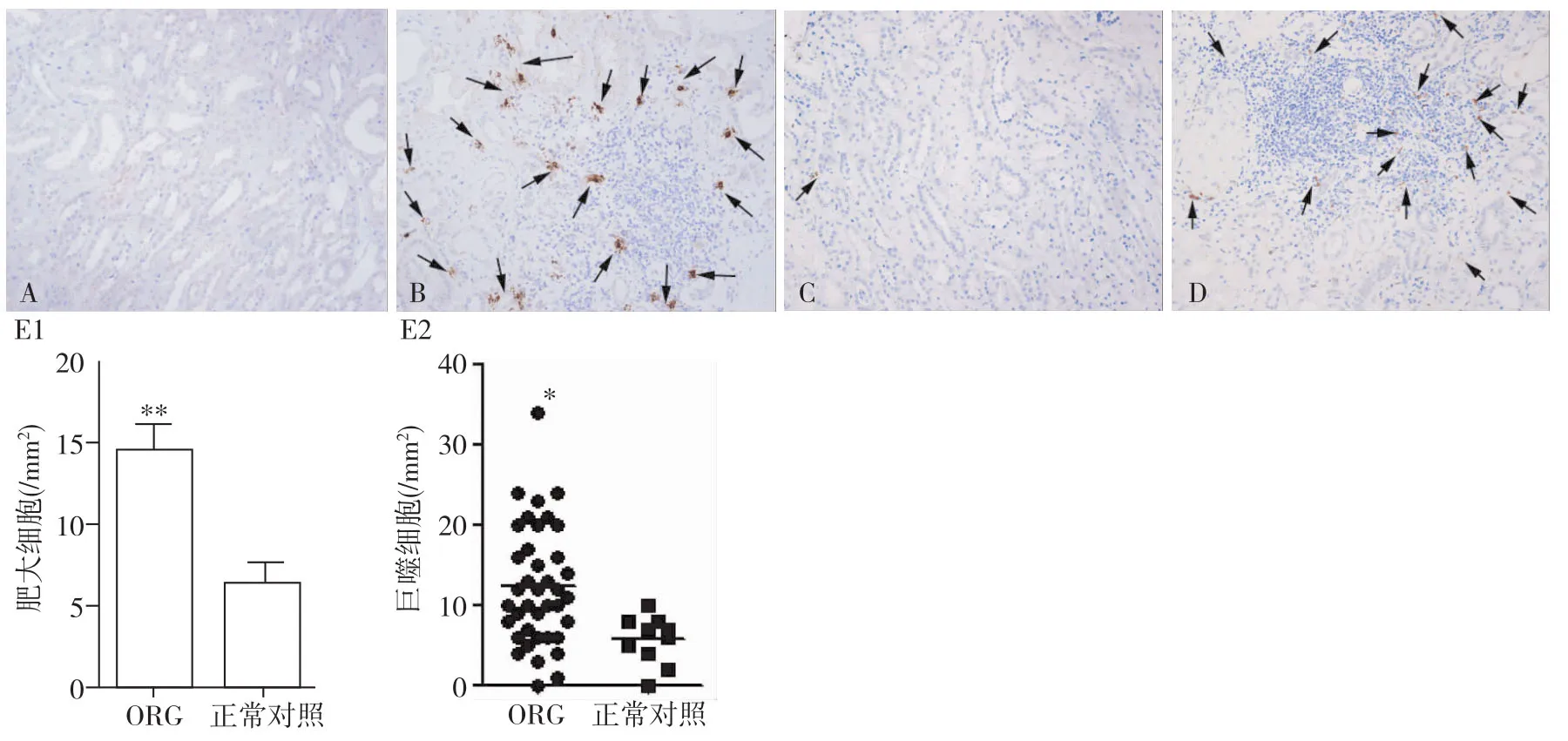
图1 肥大细胞及巨噬细胞在肥胖相关性肾病(ORG)患者肾组织中的分布(IH,×200)
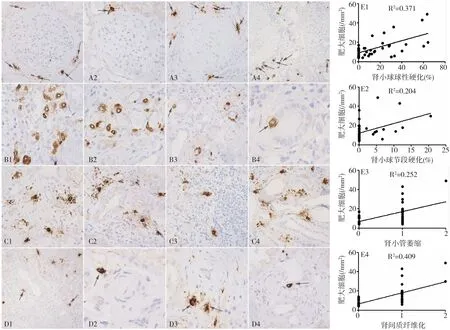
图2 肥胖相关性肾病患者肾组织肥大细胞分布特点及其与病理指标的相关性
肥大细胞与临床病理指标的关系 Pearson相关分析显示肥大细胞数量与 BMI(r=0.364,P<0.05)、血压(收缩压:r=0.459,P<0.01,舒张压:r=0.347,P<0.05)、肾小管损伤指标 NAG(r=0.324,P<0.05)以及肾功能指标(SCr:r=0.637,P<0.01,eGFR:r=-0.559,P<0.01)呈相关性(表1)。Spearman相关分析提示肥大细胞数与肾小球球性硬化(r=0.409,P<0.01)、节段硬化(r=0.457,P<0.01)、肾小管萎缩(r=0.470,P<0.01)和肾间质纤维化(r=0.669,P<0.01)呈显著正相关(图2),而与间质炎细胞数量及小动脉透明变性无明显相关性(P>0.05)。肾活检病理表现为FSGS的患者其肥大细胞数量高于单纯肾小球肥大的患者[16(6~49)vs10(4~36),P<0.05]。本研究未发现肥大细胞数量与性别、年龄、肥胖病程、空腹血糖、总胆固醇、三酰甘油、尿蛋白、RBP、IgE及CRP水平等指标的相关性(表1)。
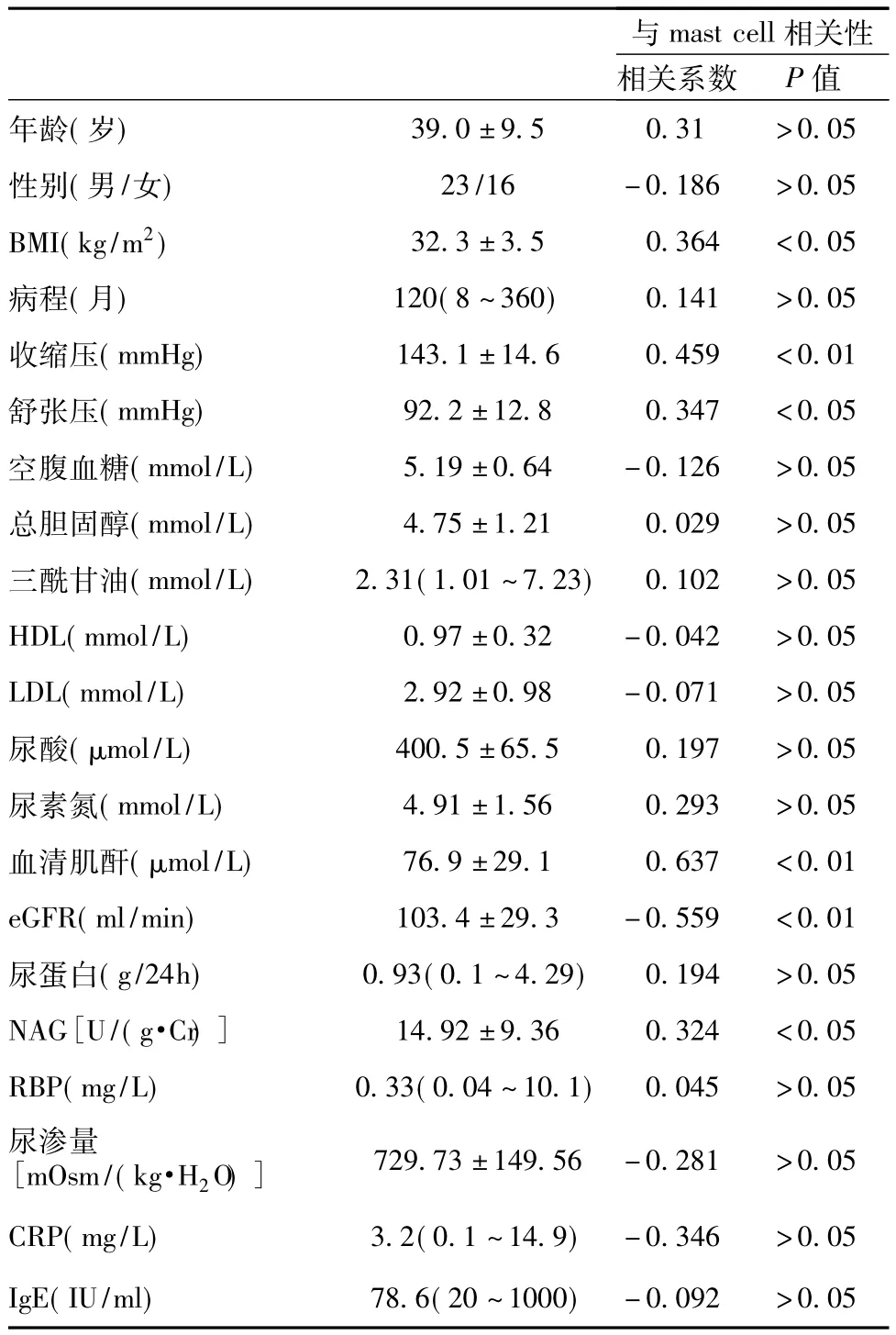
表1 肥胖相关性肾病患者肾组织肥大细胞与临床指标的关系
为探讨肾功能的影响因素,对与eGFR存在相关性的临床和病理指标进行多元回归分析,提示肥大细胞数量是影响eGFR的独立因素(R2=0.44,P<0.05)。
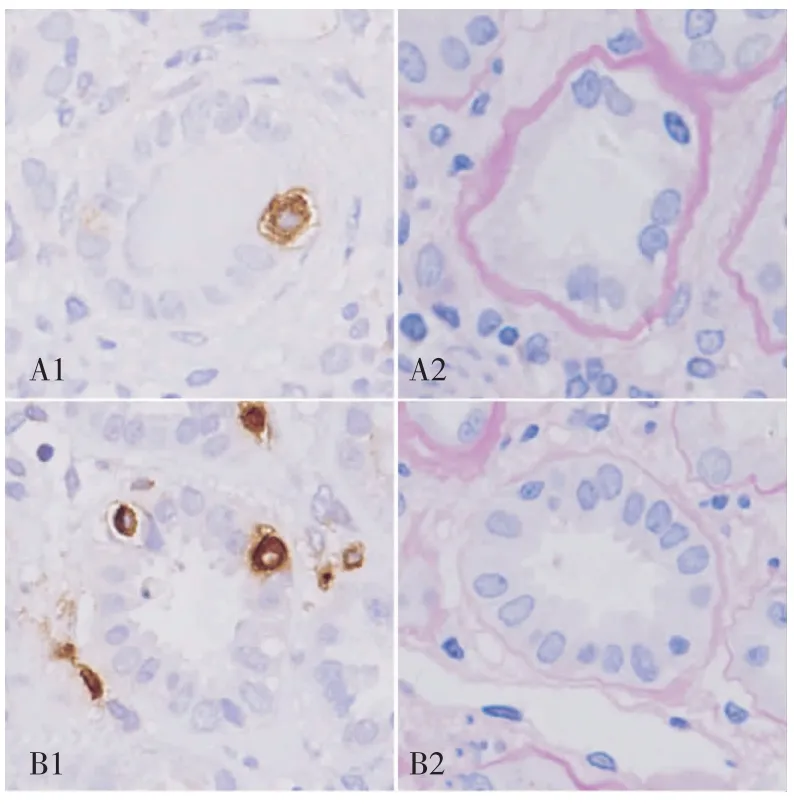
图3 肥胖相关性肾病患者肾组织肥大细胞与肾小管炎(IH,×800)
巨噬细胞分布特点及其与临床病理指标的关系 ORG肾间质巨噬细胞浸润也显著高于对照组,[11(1~34)vs6(1~10),P<0.05](图1)。巨噬细胞主要分布在有炎性细胞浸润的肾间质中,肾小球内也有少量分布。肾组织tryptase与CD68双重免疫荧光染色见肥大细胞与巨噬细胞在肾间质中的分布部位一致(图4)。
Spearman相关分析提示肾间质巨噬细胞数量与间质炎细胞浸润程度(r=0.476,P<0.01)呈正相关,而与BMI、收缩压、空腹血糖、总胆固醇、三酰甘油、NAG、RBP、肾小管萎缩、肾间质纤维化程度及肥大细胞数量等无明显相关性。
讨 论
ORG起病相对隐匿,临床上以轻、中度蛋白尿为主要表现,病理上主要表现为肾小球体积增大,伴或不伴FSGS样病变,同时伴有肾小管功能异常[13]。有研究报道ORG 5年肾存活率为77%,10年仅为51%[14]。既往研究认为降低蛋白尿是延缓ORG肾功能进展的主要措施之一,而体重控制可以显著降低ORG患者的蛋白尿水平。但预后分析证实,肾小管间质的损伤程度,而非体重及尿蛋白水平,是决定肾功能进展的重要因素[5]。然而目前关于ORG肾小管间质损伤机制仍不是很清楚。

图4 肥胖相关性肾病患者肾组织肥大细胞与巨噬细胞的关系
肥大细胞起源于多能造血祖细胞,进入组织中完成其分化,在过敏反应中发挥重要功能。但近期也有研究证实肥大细胞参与了肥胖的发生发展过程[11]。肥胖患者脂肪组织肥大细胞数量较正常人明显升高,而肥大细胞敲除小鼠其体重及体内炎性因子水平及脂肪组织巨噬细胞数量明显下降,同时,肥大细胞也参与了白色脂肪组织蛋白酶的表达及微血管的形成。但肥大细胞在ORG中的作用目前尚无相关研究。
本研究发现ORG患者肾组织肥大细胞浸润数量高于正常人,并与BMI水平呈正相关。白色脂肪组织中的定向造血干细胞可以分化为有功能的肥大细胞并迁移到不同组织[15]。而肥胖过程中肾组织糖基化终产物(advanced glycation end products,AGEs)和脂质沉积、活性氧(reactive oxygen species,ROS)产生以及过敏毒素 C3a 受体表达上调[2,16],其中C3a能够趋化肥大细胞,而AGEs、ROS及LDL均能使肥大细胞激活[17]。因此肥胖个体可能通过上述途径引起了肾组织肥大细胞的浸润及激活。肥大细胞与血压水平也呈正相关性,高血压肾损害患者的肾组织肥大细胞浸润也显著升高,伴随干细胞因子(stem cell factor,SCF)表达上调[18]。SCF 在正常肾组织表达微弱,但在疾病过程中表达上调。作为肥大细胞迁移和分化的主要刺激因子,SCF可能在肥大细胞向肾组织浸润过程中也发挥着重要作用。
肥大细胞主要在ORG肾间质纤维化处以及肾小球球囊周围分布,可见较多脱颗粒的肥大细胞,其数量与肾小球球性硬化、节段硬化和间质纤维化程度呈正相关,提示肥大细胞可能参与了肾脏纤维化过程。肥大细胞能够合成多种细胞因子、趋化因子和白三烯,可以招募并激活中性粒细胞[19];脱颗粒释放的组胺、TNF-α和转化生长因子β(transforming growth factor-β,TGF-β)能够促进纤维细胞合成胶原成分;肝素和tryptase可以引起成纤维细胞的迁移及增生[20];类糜蛋白酶(chymase)还可以影响基质金属蛋白酶/金属蛋白酶组织型抑制剂(MMPs/TIMPs)的平衡而调控细胞外基质成分沉积[21,22]。另外肥大细胞也影响肾脏局部RAS系统。肥大细胞能够合成肾素[23],脱颗粒过程中释放的肾素可以转化为血管紧张素Ⅰ(angiotensinⅠ,AngⅠ)[24]。并且chymase可以在不依赖血管紧张素转换酶(angiotensin converting enzyme,ACE)的情况下将AngⅠ转化为血管紧张素Ⅱ(angiotensinⅡ,AngⅡ)[25],体内实验证实ACEI治疗可以逆转肥大细胞引起的肾间质纤维化并且减少肥大细胞浸润数量[26]。以上均表明,肥大细胞具有促进肾脏纤维化的潜能。
本研究发现肥大细胞可以引起肾小管炎,其数量与肾小管萎缩程度呈正相关。本研究入组时排除了合并药物过敏及感染的患者,并且肾组织肥大细胞数量与血清IgE及CRP等炎性因子水平未见明显相关性,表明ORG患者肥大细胞引起肾小管炎的机制与过敏及感染引起的间质性肾炎有所不同。肾小管炎可以出现在多种非间质性肾炎中,如在移植肾,其出现提示急性排斥,肾脏预后不佳,而在糖尿病肾病及IgA肾病中,肾小管炎与肾间质炎性细胞浸润及间质纤维化有直接联系[27]。因此肾小管炎的出现与肾小管间质损伤有着重要联系,而肥大细胞参与了这一过程,进一步证实了炎症在ORG肾损伤中的作用。
巨噬细胞在肾间质纤维化中作用的研究比较广泛。经典途径激活的M1型巨噬细胞可以分泌TGF-β、MMP-9和TNF-α等引起间质纤维化,也可以通过血管损伤和组织缺氧引起肾小管萎缩[28]。而肥胖小鼠脂肪组织中,巨噬细胞可以被肥大细胞招募并分泌炎症因子引起胰岛素抵抗[11]。本研究发现巨噬细胞与肥大细胞在肾间质中的分布有一定的一致性,其数量与间质炎细胞浸润程度呈正相关,但未能明确其与肥大细胞数量及间质纤维化的关系。因此ORG肾间质损伤中巨噬细胞与肥大细胞相互作用的关系及其在组织损伤中的作用机制有待进一步阐明。
综上所述,ORG患者肾间质肥大细胞浸润数量明显增多,其程度与BMI、血压、肾间质损伤及肾功能进展之间存在显著相关性。进一步阐明肥大细胞在ORG患者肾间质损伤及肾功能进展中的作用,探索有针对性的干预措施,将有助提高ORG的诊治水平。
1 Gu D,Reynolds K,Wu X,et al.Prevalence of the metabolic syndrome and overweight among adults in China.Lancet,2005,365(9468):1398-1405.
2 Praga M,Morales E.Obesity,proteinuria and progression of renal failure.Curr Opin Nephrol Hypertens,2006,15(5):481-486.
3 Chen HM,Li SJ,Chen HP,et al.Obesity-related glomerulopathy in China:a case series of 90 patients.Am J Kidney Dis,2008,52(1):58-65.
4 Chen HM,Liu ZH,Zeng CH,et al.Podocyte lesions in patients with obesity-related glomerulopathy.Am J Kidney Dis,2006,48(5):772-779.
5 Shen WW,Chen HM,Chen H,et al.Obesity-related glomerulopathy:body mass index and proteinuria.Clin J Am Soc Nephrol,2010,5(8):1401-1409.
6 Wang Z,Jiang T,Li J,et al.Regulation of renal lipid metabolism,lipid accumulation,and glomerulosclerosis in FVBdb/db mice with type 2 diabetes.Diabetes,2005,54(8):2328-2335.
7 Ruggiero C,Ehrenshaft M,Cleland E,et al.High fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production.Am J Physiol Endocrinol Metab,2011,300(6):E1047-1058.
8 Wu Y,Liu Z,Xiang Z,et al.Obesity-related glomerulopathy:insights from gene expression profiles of the glomeruli derived from renal biopsy samples.Endocrinology,2006,147(1):44-50.
9 Olefsky JM,Glass CK.Macrophages,inflammation,andinsulin resistance.Annu Rev Physiol,2010,72:219-246.
10 Xu H,Barnes GT,Yang Q,et al.Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance.J Clin Invest,2003,112(12):1821-1830.
11 Liu J,Divoux A,Sun J,et al.Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice.Nat Med,2009,15(8):940-945.
12 Schwartz LB,Bradford TR.Regulation of tryptase from human lung mast cells by heparin.Stabilization of the active tetramer.J Biol Chem,1986,261(16):7372-7379.
13 Kambham N,Markowitz GS,ValeriAM,etal.Obesity-related glomerulopathy:an emerging epidemic.Kidney Int,2001,59(4):1498-1509.
14 Praga M,Hernandez E,Morales E,et al.Clinical features and longterm outcome of obesity-associated focal segmental glomerulosclerosis.Nephrol Dial Transplant,2001,16(9):1790-1798.
15 Poglio S,De Toni-Costes F,Arnaud E,et al.Adipose tissue as a dedicated reservoir of functional mast cell progenitors.Stem Cells,2010,28(11):2065-2072.
16 Mamane Y,Chung Chan C,Lavallee G,et al.The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation.Diabetes,2009,58(9):2006-2017.
17 Theoharides TC,Sismanopoulos N,Delivanis DA,et al.Mast cells squeeze the heart and stretch the gird:Their role in atherosclerosis and obesity.Trends Pharmacol Sci,2011.
18 Welker P,Kramer S,Groneberg DA,et al.Increased mast cell number in human hypertensive nephropathy.Am J Physiol Renal Physiol,2008,295(4):F1103-1109.
19 Benoist C,Mathis D.Mast cells in autoimmune disease.Nature,2002,420(6917):875-878.
20 Blank U,Essig M,Scandiuzzi L,et al.Mast cells and inflammatory kidney disease.Immunol Rev,2007,217:79-95.
21 Tchougounova E,Lundequist A,Fajardo I,et al.A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and promatrix metalloprotease-2.J Biol Chem,2005,280(10):9291-9296.
22 Frank BT,Rossall JC,Caughey GH,et al.Mast cell tissue inhibitor of metalloproteinase-1 is cleaved and inactivated extracellularly by alphachymase.J Immunol,2001,166(4):2783-2792.
23 Silver RB,Reid AC,Mackins CJ,et al.Mast cells:a unique source of renin.Proc Natl Acad Sci USA,2004,101(37):13607-13612.
24 Mackins CJ,Kano S,Seyedi N,et al.Cardiac mast cell-derived renin promotes local angiotensin formation,norepinephrine release,and arrhythmias in ischemia/reperfusion.J Clin Invest,2006,116(4):1063-1070.
25 Huang XR,Chen WY,Truong LD,et al.Chymase is upregulated in diabetic nephropathy:implications for an alternative pathway of angiotensin II-mediated diabetic renal and vascular disease.J Am Soc Nephrol,2003,14(7):1738-1747.
26 Jones SE,Kelly DJ,Cox AJ,et al.Mast cell infiltration and chemokine expression in progressive renal disease.Kidney Int,2003,64(3):906-913.
27姜傥关,周建中.肾小管炎在肾小球性疾病中的临床意义.中山大学学报(医学科学版),2003,24(1):63-67.
28 Vernon MA,Mylonas KJ,Hughes J.Macrophages and renal fibrosis.Semin Nephrol,2010,30(3):302-317.
猜你喜欢
昆明医科大学学报(2021年12期)2021-12-30 06:59:44
中国民间疗法(2021年19期)2021-11-20 06:22:34
中国民间疗法(2021年18期)2021-11-02 08:20:16
中国兽医杂志(2018年5期)2018-08-24 03:16:34
中国继续医学教育(2015年2期)2016-01-06 01:36:29
中外医疗(2015年11期)2016-01-04 03:58:45
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
西南军医(2015年6期)2015-01-23 01:25:49
医学研究与教育(2014年2期)2014-03-11 17:59:25
