
C3肾小球肾炎的临床表现及病理特征
2011-08-07 08:12章海涛陈惠萍曾彩虹邓康平李世军郑春霞刘志红
肾脏病与透析肾移植杂志 2011年4期
章海涛 陈惠萍 曾彩虹 邓康平 李世军 郑春霞 刘志红
C3肾小球肾炎指免疫荧光染色补体C3沿肾小球毛细血管袢沉积,不伴其他免疫球蛋白沉积为特征,电镜观察见肾小球毛细血管袢内皮下和(或)系膜区电子致密物沉积的肾小球肾炎[1,2]。其临床表现以水肿、蛋白尿为主,可伴镜下血尿、高血压和血清肌酐(SCr)升高,组织学改变以膜增生性肾小球肾炎(MPGN)为多,部分患者体内检测到C3肾炎因子(C3 nephritic factor,C3NeF)和补体旁路调节蛋白的变异[1]。既往常将此病与MPGN及致密物沉积病(dense deposit disease,DDD)相混淆,随着对补体调节系统的深入研究,人们逐渐认识到这是一类相对独立的疾病,其发病机制与补体调节异常密切相关[2]。虽然目前对C3肾小球肾炎的诊断已达成一定共识,但仍有一些问题需进一步商榷。本文回顾性分析17例C3肾小球肾炎的临床病理资料。
对象和方法
病例选择 2004年9月至2010年3月于南京军区南京总医院全军肾脏病研究所住院行肾活检,依临床表现、实验室检查及肾穿刺结果符合C3肾小球肾炎者,排除DDD、MPGN(Ⅰ型和Ⅲ型)、冷球蛋白血症、系统性红斑狼疮(SLE)、感染后肾小球肾炎、乙型或丙型肝炎病毒感染、甲状腺相关疾病及恶性肿瘤等。
临床观察指标 诱因,首发症状,肾外症状,肾脏损害的症状、体征,血常规、肝肾功能、24h尿蛋白定量、尿沉渣红细胞计数,血清C3NeF、补体H因子、抗H因子抗体、冷球蛋白、自身抗体及肾脏B超检查等。血清C3NeF采用绵羊红细胞溶血法,正常参考值<10%;补体H因子采用ELISA法(商品化试剂盒,Hycult公司)检测,正常人群范围:203~1 053 μg/ml;抗H因子抗体采用纯化H因子包被96孔酶标板,再加入辣根过氧化物酶(HRP)标记的鼠抗人IgG,显色后酶标仪450 nm读取吸光度值,正常人群范围:0.43~0.71。
临床指标定义 高血压:收缩压≥140 mmHg或舒张压≥90 mmHg;贫血:血红蛋白 <110 g/L;SCr升高:SCr>109.6 μmol/L;终末期肾病(ESRD):SCr>707.2 μmol/L,需行肾脏替代治疗。
肾活检病理 17例患者均在B超引导下行经皮肾穿刺活检术。光镜组织经10%甲醛固定、石蜡包埋后连续切片,切片厚度1.5 μm,常规行 HE、PAS、PASM和Masson三色染色;肾组织免疫荧光染色采用直接法,冰冻切片厚度 4 μm,行 IgG、IgA、IgM、C3、C4、C1q和 Fibrin染色,观察免疫球蛋白和补体沉积的部位和强度;电镜组织以3.75%冷戍二醛固定,1%四氧化锇后固定,70~80 nm超薄切片,醋酸铀、柠檬酸铅双染色,置Hitachi 7500透射电子显微镜下观察。
疗效判断 完全缓解(complete remission,CR):尿蛋白定量恢复正常(<0.4 g/d),尿沉渣红细胞计数≤20万/ml,血清白蛋白≥35 g/L、SCr在正常范围;部分缓解(partial remission,PR):治疗后蛋白尿定量下降超过基础值50%且<3.5 g/d,尿沉渣红细胞计数下降超过基础值50%、SCr正常或上升不超过30%、血清白蛋白≥30 g/L。
统计学方法 采用SPSS 11.0统计软件进行数据处理,计量资料采用均数(标准差表示,计数资料采用病例数和(或)百分数描述。
结 果
一般资料 17例患者中男11例,女6例;年龄9~72岁,平均(31.5±19.4)岁;病程1周 ~8年,平均(30.0±27.8)月。
诱因及首发症状 14例患者起病前无明确诱因,上呼吸道感染和腹泻各1例,1例曾接触花粉。水肿、尿检异常为常见的首发症状13例(76.5%),3例伴肾外症状(皮肤、关节症状)。
肾脏损害临床表现及实验室检查 17例患者中5例肾病综合征,8例尿检异常,3例急进性肾炎,1例反复发作肉眼血尿。尿蛋白定量平均(3.1±3.2)g/d,大量蛋白尿(>3.5 g/d)6例(尿蛋白定量波动在3.6~13.5 g/d),血清白蛋白(33.8±6.1)g/L。镜下血尿(尿沉渣红细胞计数>20万/ml)11例(64.7%),肉眼血尿4例(23.5%)、高血压10例(58.8%)、SCr升高4例(23.5%)。补体 C3下降11例(64.7%),其中3例补体C3<0.2 g/L,贫血7例。5例患者检测了C3NeF、补体H因子及抗H因子抗体,其中1例C3NeF阳性(表1)。

表1 17例C3肾小球肾炎患者肾活检时的临床及实验室资料
肾脏病理 肾活检免疫荧光染色证实全部患者补体C3呈颗粒状弥漫分布于肾小球毛细血管外周袢(图1),其中8例仅见C3沉积,另9例同时合并节段、非特异性IgM沉积(图2)。肾活检组织学符合膜增生样病变12例,肾小球体积增大,弥漫系膜细胞、内皮细胞增生,球内浸润细胞(单核为主,少数中性粒细胞)(图3),1例新月体形成。特殊染色节段肾小球外周袢分层,见双轨形成,7例光镜下见系膜区、内皮下嗜复红物(图4),偶见上皮侧嗜复红物沉积。以肾小球系膜增生病变为特征者5例,其中3例肾小球系膜重度增生,2例除系膜增生外,还伴肾小球内皮细胞增生。3例患者球性废弃达45%~55%,肾小管中度萎缩8例(47.1%),中度纤维化2例(11.8%),小动脉内膜增厚分层5例,透明变性5例,1例纤维素样变性。17例患者电镜观察见肾小球毛细血管袢内皮下电子致密物沉积(图5,6),呈团块状或散在、颗粒状,系膜区可见电子致密物,8例患者偶见上皮侧电子致密物沉积,其周见基膜样物质,7例患者基膜内偶见电子致密物沉积,7例患者内皮下疏松、区域增宽;3例肾小球内皮细胞病变突出;广泛足突融合,较多微绒毛化,胞浆内见扩张的内质网、空泡、高电子密度的溶酶体及吞噬性溶酶体。
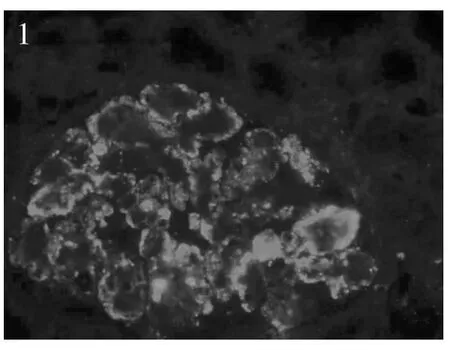
图1 C3弥漫分布于肾小球外周袢(IF,×400)
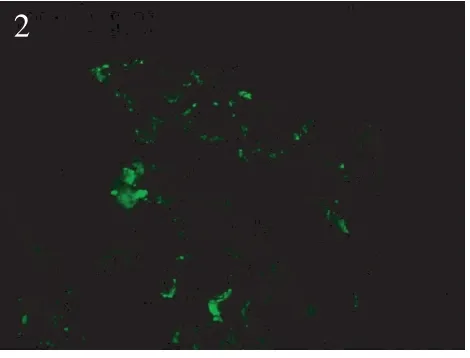
图2 IgM非特异性沉积(IF,×400)
随访治疗 5例患者(均为重复肾活检患者)入院前曾接受大剂量免疫抑抑制剂治疗,但效果不佳。17例患者中,除1例失访外,余16例患者随访观察4月~5年(表2),多数患者采用雷公藤多苷及免疫抑制剂治疗。至随访观察终点,尿检1例CR,6例PR,7例病情无变化,1例 SCr倍增,1例进入ESRD。

图3 肾小球积增大,分叶状,弥漫系膜、内皮细胞增生,袢内单个核及中性粒细胞浸润(PAS,×400)
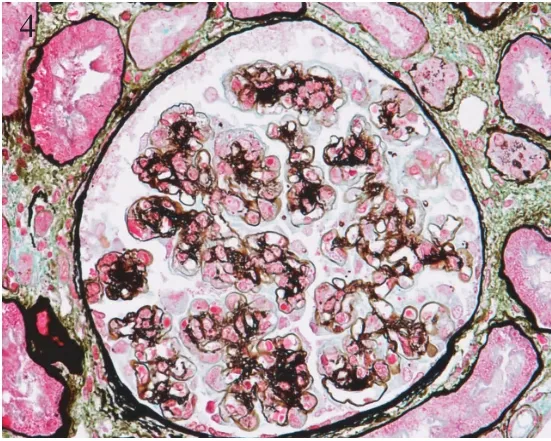
图4 肾小球毛细血管袢内皮下及系膜区嗜复红物沉积,节段外周袢分层(PASM-Masson,×400)
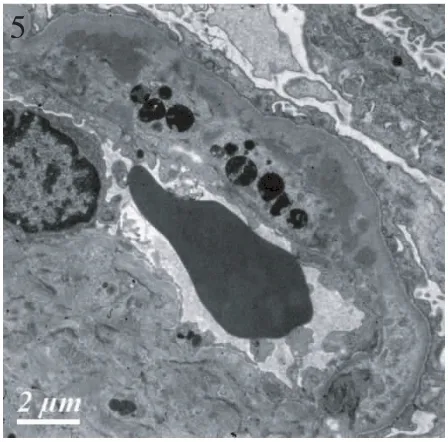
图5 肾小球毛细血管袢内皮下电子致密物沉积,系膜细胞胞质插入至内皮下(EM)
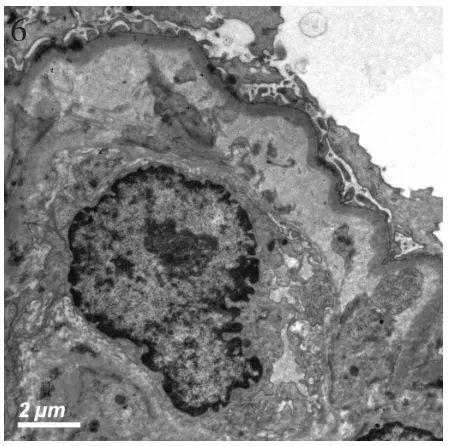
图6 肾小球毛细血管袢内皮下疏松、区域增宽,其中见细胞碎屑(EM)
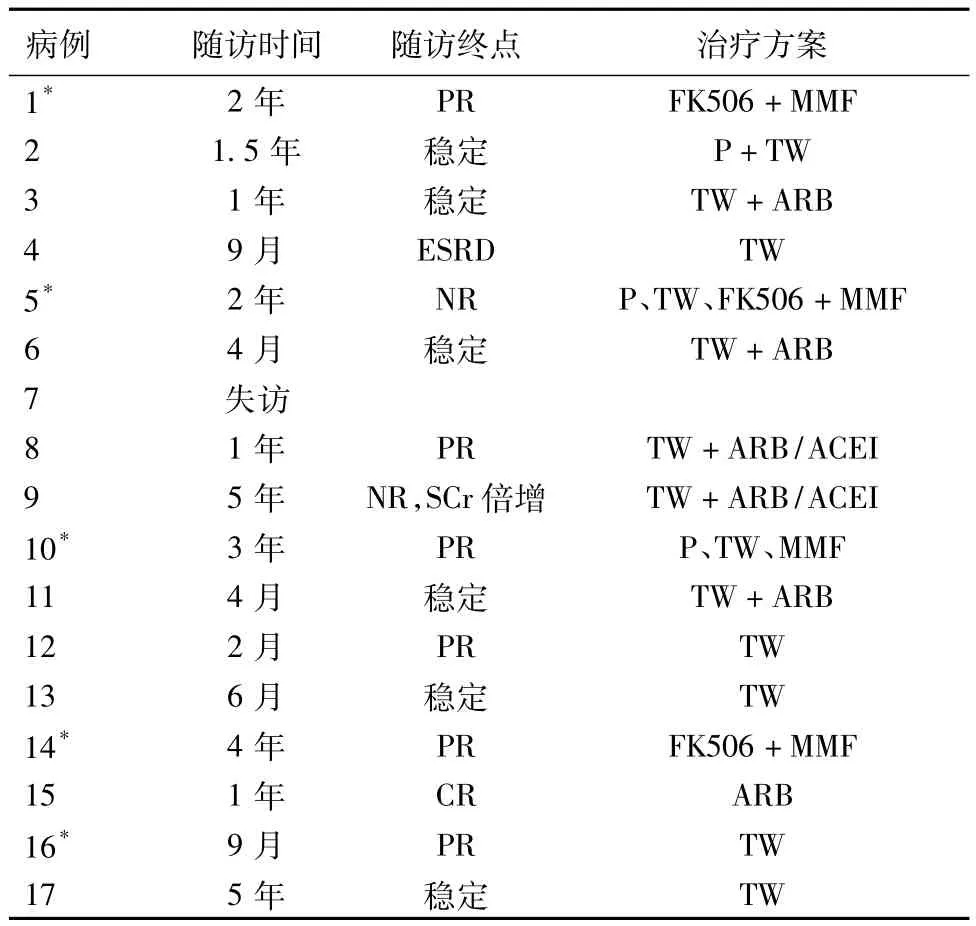
表2 17例C3肾小球肾炎患者的随访情况
讨 论
定义及分类 许多肾小球疾病的发病机制与补体系统调节异常相关[3-5],如溶血尿毒综合征和DDD 等[6,7],随着对补体调节系统的深入了解,人们重新认识了这类与补体调节异常相关的疾病。
近年来,将一组以免疫荧光染色补体C3沿肾小球毛细血管袢沉积为主的肾小球疾病统一命名为C3肾病(C3 glomerulopathy)或 C3沉积病(C3 deposition glomerulopathy,C3 DG)[1,2]。这组疾病除免疫荧光染色以补体C3沿肾小球毛细血管袢沉积为主外,发病机制均与补体系统调节异常有关[1,2],尽管其具有不同的组织学改变和临床表现,但均统称“C3肾病”。
DDD是C3肾病中认识较深入、不易误诊的疾病。典型的DDD具有特殊的组织学及超微结构特征[6,8-10],其发病机制与免疫复合物无关,而与体内补体系统调节异常(如抗补体H因子抗体、C3NeF及补体H因子遗传缺陷等)相关[6-9]。因此,人们将其与Ⅱ型MPGN区分,认为是一类单独的疾病。
C3肾病中还包括家族性Ⅲ型MPGN、CFHR5肾病及单纯补体C3沉积的I型MPGN,它们在发病机制上也与补体调节异常相关[11-13]。另一类缺乏免疫球蛋白沉积,仅有补体C3沉积的肾炎[2],人们将其命名为“C3 肾小球肾炎”[1,2](表3)。

表3 C3肾病的诊断标准及分类
病理特征 C3肾小球肾炎依靠免疫病理确诊[1,2],其免疫荧光染色特点及超微结构改变具有特征性,不同于Ⅰ型MPGN和DDD(表4)。
本组17例患者除C3沿肾小球外周袢分布外,9例见非特异性IgM沉积。IgM分子量较大,极易滞留于病变的肾小球节段,但不具致病性,亦无临床意义。

表4 C3肾小球肾炎与DDD、特发性Ⅰ型MPGN的鉴别[2]
文献报道C3肾小球肾炎组织学改变以MPGN为多,约占75%,因此称“MPGN样C3肾炎”,此外,部分C3肾小球肾炎免疫荧光染色以肾小球系膜区和上皮侧C3沉积,组织学不符合膜增生样病变,称为“非 MPGN C3 肾炎”[1]。本组患者中 12 例(70.6%)符合膜增生样病变,5例符合非MPGN C3肾炎。因此,提示对一些非膜增生样病变的患者,如免疫荧光染色表现典型的C3沿血管袢分布,应结合临床及实验室检查明确是否为C3肾小球肾炎。
超微结构观察对鉴别C3肾小球肾炎和DDD有重要价值,两者最主要的区别在电子致密物沉积的部位(表4)。C3肾小球肾炎以内皮下和(或)系膜区电子致密物沉积为主,而DDD则以电子致密物沉积在肾小球基膜,尤其致密层内为特征[1,2,10]。
本文报道的17例患者均见肾小球毛细血管内皮下电子致密物沉积,其中3例患者内皮细胞病变明显,肾小球内皮细胞病变是否与C3肾小球肾炎有关,还有待深入研究。此外,我们也观察到8例患者偶见肾小球基膜上皮侧电子致密物沉积,7例肾小球基膜内电子致密物沉积。
临床表现及实验室检查特点 对C3肾小球肾炎的临床研究相对少[1,2]。文献报道其临床表现缺乏特征性,与MPGN类似;常以肾病综合征,伴血尿、高血压及肾功能损害为主要表现,多存在补体降低[1]。本文报道的患者中肾病综合征 5例(29.4%),尿检异常 8例(47.1%),3例(17.6%)以急进性肾炎起病。血尿、高血压、肾功能损害的发生率分别是64.9%,58.8%,23.5%。2/3患者存在低补体血症。
由于C3肾小球肾炎的发病机制与补体调系统有关,因此补体因子及补体调节因子突变的检测有重要意义[4,8,11,13,14]。补体旁路途径激活因子( 如C3NeF)或阻止补体旁路途径调节蛋白的活化(如抗补体H因子抗体)或变异可导致补体旁路途径异常[1,4]。多数患者是由于补体调节因子丢失,如C3NeF,也有少数是补体 H因子突变导致[7]。Servais等[1]研究发现MPGN样C3肾炎中C3NeF阳性率高于补体 H因子、I因子突变(38.5%vs15.3%),而在非MPGN C3肾炎中则相反,分别为16.7%和66.7%。遗憾的是我们仅检测了5例患者的C3NeF、H因子及抗H因子抗体,仅1例患者C3NeF阳性且组织学表现为膜增生样病变。由于本文属于回顾性分析,多数患者未能检测补体相关因子,有待今后重点评估。
治疗及预后 对C3肾小球肾炎治疗和预后观察的报道较少。文献报道50%患者持续肾功能正常,但也有15.8%患者可进展为ESRD[1]。有学者认为其预后与DDD相似。
目前缺乏对C3肾小球肾炎治疗的有效手段,多数学者仍建议免疫抑制剂治疗。本文中5例重复肾活检的患者都曾使用过大剂量免疫抑制剂,然疗效欠佳。我们观察的患者中16例采用雷公藤多苷及免疫抑制剂治疗,随访4月~5年,7例有效,另有7例病情无变化,仅2例进入肾功能不全。目前总体预后尚好,不同组织学改变是否与预后相关尚不确切。由于病例数少,还需继续随访。
小结:C3肾小球肾炎是一种新近被认可的疾病,其发生机制与补体系统调节异常密切相关。缺乏特征性临床表现,免疫荧光染色C3沿肾小球毛细血管袢沉积,超微结构改变以肾小球毛细血管袢内皮下和(或)系膜区电子致密物沉积为特征,组织学改变以膜增生样病变为多,目前尚无有效治疗方法,我们应重视此病的诊断。
1 Servais A,Freémeaux-BacchiV,LequintrecM,etal.Primary glomerulonephritis with isolated C3 deposits:a new entity which sharescommon genetic risk factors with haemolytic uraemic syndrome.J Med Genet,2007,44(3):193-199.
2 Fakhouri F,Frémeaux-Bacchi V,Noël LH,et al.C3 glomerulopathy:a new classification.Nat Rev Nephrol,2010,6(8):494-499.
3 Pickering MC,Cook HT.Translationalmini-review series on complement factor H:renal diseases associated with complement factor H:novel insights from humans and animals.Clin Exp Immunol,2008,151(2):210-230.
4 Rose KL,Paixao-Cavalcante D,Fish J,et al.Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice.J Clin Invest,2008,118(2):608-618.
5 Welch TR.The complement system in renal diseases.Nephron,2001,88(3):199-204.
6 Appel GB,Cook HT,Hageman G,et al.Membranoproliferative glomerulonephritis type II(dense deposit disease):an update.J Am Soc Nephrol,2005,16(5):1392-1403.
7 Pickering M,Cook HT.Complement and glomerular disease:new insights.Curr Opin Nephrol Hypertens,2011,20(3):271-277.
8 Walker PD,Ferrario F,Joh K,et al.Dense deposit disease is not a membranoproliferative glomerulonephritis.Mod Pathol,2007,20(6):605-616.
9 Smith RJ,Alexander J,Barlow PN,et al.New approaches to the treatment of dense deposit disease.J Am Soc Nephrol,2007,18(9):2447-2456.
10甄军晖,孙海平.膜增生肾性肾小球肾炎和致密物沉积病//黎磊石,刘志红.中国肾脏病学.北京:人民军医出版社,2008:402-421.
11 Licht C,Fremeaux-Bacchi V.Hereditary and acquired complement dysregulation in membranoproliferative glomerulonephritis.Thromb Haemost,2009,101(2):271-278.
12 Neary JJ,Conlon PJ,Croke D,et al.Linkage of a gene causing familial membranoproliferative glomerulonephritis type III to chromosome 1.J Am Soc Nephrol,2002,13(8):2052-2057.
13 Gale DP,de Jorge EG,Cook HT,et al.Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis.Lancet,2010,376(9743):794-801.
14 Habbig S,Mihatsch MJ,Heinen S,et al.C3 deposition glomerulopathy due to a functional factor H defect..Kidney Int,2009,75(11):1230-1234.
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年2期)2021-03-29
肝博士(2021年1期)2021-03-29
中国医药指南(2017年3期)2017-11-13
成都中医药大学学报(教育科学版)(2016年1期)2016-01-22
中华皮肤科杂志(2014年3期)2014-12-19
中国火炬(2014年8期)2014-07-24
实验动物与比较医学(2014年6期)2014-02-28
成都中医药大学学报(教育科学版)(2014年1期)2014-01-19
