
高效液相色谱法同时测定人血浆中多种喹诺酮类药物浓度
2011-08-06 02:28温中明徐玉红
中国药业 2011年14期
温中明,李 东,徐玉红
(1.广东省深圳市光明新区公明人民医院药剂科,广东 深圳 518106;2.暨南大学第二临床医学院·深圳市人民医院临床药学室,广东 深圳 518020;3.广东医学院附属深圳福田人民医院药剂科,广东 深圳 518033)
喹诺酮类药物的杀菌效果呈浓度依赖性,浓度越高,抗菌活性越强,最大血药浓度与最低抑菌浓度的比值(Cmax/MIC)大于8~10时,呈明显杀菌活性,且可减少耐药菌株的产生。但随着浓度增加,药物不良反应明显增加,对于某些特殊患者,抗菌药物的血药浓度监测具有明显的临床意义。有关血样中喹诺酮类药物的含量测定方法报道很多[1-8],但测定不同的喹诺酮类药物,文献报道所使用的仪器、色谱条件以及样品处理方式各不相同,有些方法对实验设备或技术要求较高,基层实验室难以满足。为此,笔者建立了一种血样处理相对简单、条件易于满足、检测灵敏度高的方法,可用于多种喹诺酮类药物的测定,现报道如下。
1 仪器与试药
Agilent 1100型高效液相色谱仪,包括G 1311 A四元泵,G 1322 A真空脱气机,G 1315 B-DAD检测器,D 1316 A柱温箱,rheodyne-7725i进样器,ChemStation色谱工作站;AG 285型电子天平(梅特勒-托利多仪器有限公司);XW-80A型旋涡混合器(上海医科大学仪器厂);KQ 5200 DB型超声波清洗器(昆山市超声仪器有限公司);高速离心机(雅培公司);pHS-3C数字pH计(上海精科雷磁仪器厂)。氧氟沙星对照品、洛美沙星对照品、加替沙星对照品、司帕沙星对照品均购自中国药品生物制品检定所;盐酸莫西沙星对照品由拜耳医药保健有限公司提供;甲醇、乙腈为一级色谱纯,均购自天津四友精细化学品有限公司;水为注射用水,其他试剂为分析纯。所有试液使用前均经0.45 μm微孔滤膜过滤。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Kromasil C8柱(250 mm ×4.6 mm,5 μm);流动相:0.02 mol/L磷酸二氢钾缓冲液(调pH至3.0)-甲醇-乙腈 (65∶20 ∶15);流速:1.0 mL/min;柱温:30 ℃;进样量:20 μL。检测波长依测定物进行调整以提高检测灵敏度,检测模型药物莫西沙星时波长设定为296 nm。在此条件下,氧氟沙星、洛美沙星、加替沙星、司帕沙星、莫西沙星均能依次得到分离,其中加替沙星和莫西沙星的保留时间分别约为5.8 min和7.6 min,理论板数按莫西沙星计算不低于8 000。色谱图见图1。

图1 人血浆中5种喹诺酮类抗菌药物高效液相色谱图
2.2 溶液配制
精密称取干燥至恒重的各对照品10.0~20.0 mg,置10 mL量瓶中,以注射用水溶解并稀释至刻度,摇匀,分别得质量浓度为1 000.0~2 000.0 μg/mL 的对照品贮备液,置 4 ℃冰箱中备用。莫西沙星对照品贮备液的质量浓度为1 000.0 μg/mL,临用时分别精密量取适量于10 mL量瓶中,以流动相稀释至刻度,得质量浓度分别为 2.5,5,10,20,40,80,160,250 μg/mL 的系列莫西沙星对照品工作液。加替沙星对照品贮备液的质量浓度为1 000.0 μg/mL,作内标时的质量浓度为20.0 μg/mL。精密称取磷酸二氢钾2.72 g,加适量蒸馏水溶解后加入10 mL三乙胺,边加边搅拌,以蒸馏水定容至1 000 mL,用分析纯浓磷酸调 pH至3.0,即得浓度为0.02 mol/L的磷酸盐缓冲液。
2.3 血浆样品预处理
将全血样品4 mL置肝素抗凝管中,静置30 min,以3 000 r/min离心10 min。取上清液,置1.5 mL离心管中,密封,标记,-20℃保存至分析。分析前,血浆样品于室温下自然解冻后,精密吸取0.5 mL,置10 mL具塞锥形离心管中,加内标溶液适量,旋涡混合30 s,加入二氯甲烷5 mL,旋涡提取5 min,10 000 r/min离心10 min,取二氯甲烷层,50℃水浴氮气流吹干,残渣以100 μL流动相旋涡30 s溶解,再以10 000 r/min离心3 min后取20 μL进样。以加替沙星为内标测定模型药物莫西沙星时,20.0 μg/mL的加替沙星内标溶液的加入量为 15 μL。
2.4 方法学考察
专属性试验:分别取6名健康体检者的混合空白血浆0.5 mL,按2.3项下操作,获得空白血浆样品的典型色谱图1A;将不同质量浓度的5种喹诺酮类药物对照品溶液10 μL加入至0.5 mL混合空白血浆中,同法操作,获得典型色谱图1B。将一定质量浓度莫西沙星对照品溶液10 μL加入0.5 mL混合空白血浆中得0.2 μg/mL莫西沙星对照品血样,同法操作,获得典型色谱图1C;取健康受试者单剂量口服400 mg盐酸莫西沙星片剂后12 h的血浆样品,同法操作,得色谱图1D。结果表明,在筛选出的样品处理条件及色谱条件下,血浆中内源性物质及代谢物均不干扰样品测定,峰形对称性好,基线噪音小(图1)。
线性范围考察及检测限:以莫西沙星为模型药物,对方法学进行验证。取10 mL具塞锥形离心管9支,分别精确加入健康体检者空白血浆0.5 mL,除空白管外,每支管精确加入不同质量浓度的莫西沙星对照品工作液10 μL,涡旋混合30 s,使质量浓度分别为0.05,0.10,0.20,0.40,0.80,1.60,3.20,5.00 μg/mL,按 2.3 项下方法操作,分别记录莫西沙星峰面积(As)及内标峰面积(Ai),以莫西沙星与内标峰面积比值(As/Ai)对莫西沙星质量浓度(C)进行线性回归,得回归方程 As/Ai=2.119 4 C -0.077 9(r=0.999 8)。结果表明,莫西沙星质量浓度线性范围是 0.05~5.00 μg/mL。莫西沙星的最低定量限为 0.05 μg/mL,检测限为 0.015 μg/mL(RS/N=3)。
方法精密度:用空白血浆及对照品溶液配制质量浓度为0.10,1.60,5.00 μg/mL 的 3 种莫西沙星质控样品,各 6 份,按 2.3 项下方法操作,于1 d内各浓度重复测定6次;连续6 d,每天配制上述3种质量浓度的莫西沙星样品进行测定,将测定结果比值代入回归方程,计算日内、日间精密度。结果见表1。
表1 方法日内及日间精密度试验结果(±s,n=6)
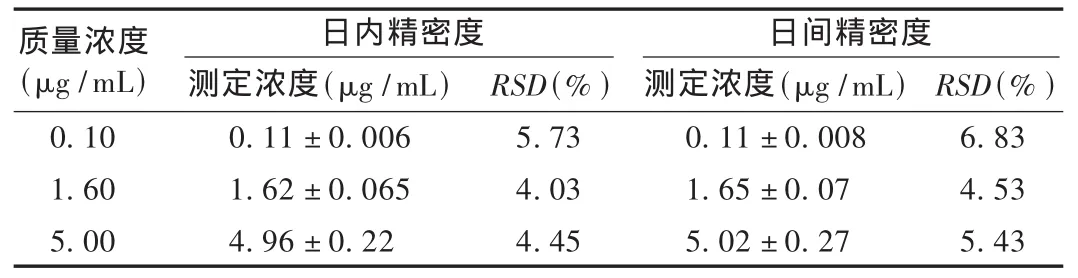
表1 方法日内及日间精密度试验结果(±s,n=6)
质量浓度(μg/mL)日内精密度 日间精密度0.10 1.60 5.00测定浓度(μg/mL)0.11 ± 0.006 1.62 ± 0.065 4.96 ± 0.22 RSD(%)5.73 4.03 4.45测定浓度(μg/mL)0.11 ± 0.008 1.65 ± 0.07 5.02 ± 0.27 RSD(%)6.83 4.53 5.43
回收率:用空白血浆0.5 mL和莫西沙星对照品溶液适量,配制质量浓度为 0.10,1.60,5.00 μg/mL 的 3 种莫西沙星质控样品,各6份,按2.3项下方法操作,测定。将莫西沙星与加替沙星峰面积比代入线性方程,计算质量浓度,测定质量浓度与加入质量浓度的比值即为方法回收率。另取空白血浆0.5 mL,不加入莫西沙星及内标工作液,按2.3项下方法处理,在取二氯甲烷层后加入相同量的莫西沙星及内标工作液,旋涡30 s,同法吹干、溶解、测定。将提取后测得的峰面积与未提取测得的峰面积进行比较,计算提取回收率。结果见表2。
表2 方法回收率和提取回收率试验结果(±s,n=6)
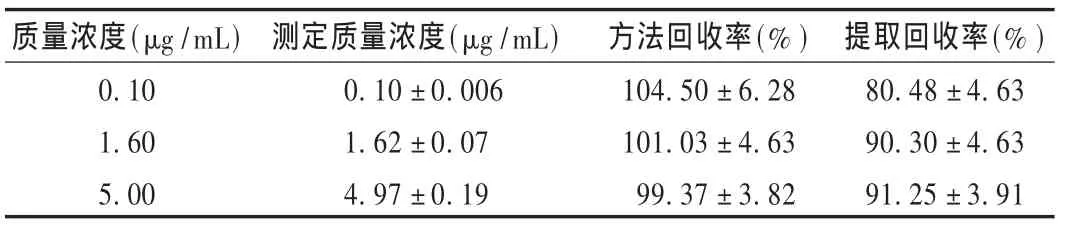
表2 方法回收率和提取回收率试验结果(±s,n=6)
质量浓度(μg/mL)0.10 1.60 5.00测定质量浓度(μg/mL)0.10 ±0.006 1.62 ±0.07 4.97 ±0.19方法回收率(%)104.50 ± 6.28 101.03 ± 4.63 99.37 ± 3.82提取回收率(%)80.48 ± 4.63 90.30 ± 4.63 91.25 ± 3.91
血浆样品稳定性:用空白血浆配制足够量的质量浓度为0.10,1.60,5.00 μg/mL 的莫西沙星样品,立即按 2.3 项下方法处理、测定。在-20℃冰箱中冷冻24 h后取出,室温解冻后,各取样0.5 mL处理、测定。剩余血浆样品再次放入-20℃冰箱中冷冻,24 h后解冻、处理、测定,重复冻融循环3次,考察血浆样品冻融稳定性。结果低、中、高质量浓度的 RSD 分别为 5.27%,4.03%,5.25% ,显示冻融条件对血浆样品的检测结果无明显影响。用空白血浆配制足量的质量浓度为 0.10,1.60,5.00 μg/mL 的莫西沙星样品,吸取各质量浓度样品1 mL,置1.5 mL离心管中,每个质量浓度9份。其中5 份在室温下放置 0,2,4,8,12 h 后按 2.3 项下方法处理、测定;另外4份分别在-20℃冰箱中保存1,2,4,12周后,再按2.3项下方法处理、测定。3个质量浓度室温下测定结果的 RSD为4.18% ~5.77%,冷冻条件下测定结果的 RSD为4.58% ~8.15%,表明莫西沙星血浆样品在室温放置8 h和-20℃冷冻条件下均有很好的稳定性。
2.5 样品分析
健康受试者(2周内未服用任何可能影响本品吸收、代谢的药物)于早晨空腹以200 mL温水送服单剂量莫西沙星400 mg,于服药后1,3,24 h抽取静脉血5 mL共3份,按拟订的方法进行处理和测定。结果3 份样品的血药浓度分别为 2.60,1.82,0.48 μg/mL,表明建立的方法可用于实际药物浓度的测定。
3 讨论
喹诺酮类药物是以4-喹诺酮(或称为吡酮酸)为基本结构的人工合成药物,在 N1,C3,C6,C7,C8位引入不同基团而形成不同药物,同时也导致其理化性质、抗菌活性有所差异,化学结构的相似性也为该类药物的同时分离提供了可能[1]。由于结构中含有羧基以及可质子化的氮原子,喹诺酮类药物均具有两性化合物的特性,色谱行为受流动相pH的影响较大。利用液相色谱进行分离时,碱性基团会与固定相未封闭的残余硅醇基之间发生相互作用,导致分离时间延长,发生拖尾现象。碱性越强,拖尾现象越严重,如采用Hypersil C18柱分离受试的几种喹诺酮类药物时,莫西沙星保留时间最长,拖尾现象最严重,灵敏度无法满足测定要求,调节pH、添加扫尾剂也未得到明显改善。将分析柱调整为C8柱后,莫西沙星峰形明显改善、灵敏度增高、保留时间适中,因此本试验中选用C8柱作为分离柱。
作为酸碱两性化合物,喹诺酮类药物在水溶液中可以多种离子形式存在,需对流动相的pH进行调节。筛选试验结果表明,pH为3.0~4.0时对大多数药物较合适,低pH流动相也可抑制硅醇的离解,有利于改善峰形;加入三乙胺可进一步改善峰形,对其使用浓度进行筛选后选用1%三乙胺。综合考虑灵敏度、保留时间和选择性,最终选择以磷酸缓冲盐、甲醇、乙腈为流动相,以三乙胺作为扫尾剂调节峰形和灵敏度。在实际检测时,可以根据分析的品种对流动相比例进行微调,以使分离效果最佳化。由于含有的取代基团不一样,受试的几种药物最大吸收波长并不一致,在进行实际样品分析时,可根据具体分析品种选择合适的检测波长,以进一步提高检测灵敏度。
试验表明,采用常用的几种蛋白沉淀试剂直接沉淀蛋白时,干扰物较多,且灵敏度较差。在一定pH条件下,以二氯甲烷提取后可消除大部分内源性干扰物质。在测定莫西沙星时,采用0.5 mol/L的氢氧化钠10 μL以调节萃取液酸碱度,可明显提高莫西沙星的萃取效率。样品测定结果表明,建立的高效液相色-紫外分光光度法可用于人体血样浓度的测定。
[1]Samanidou VF,Christodoulou EA,Papadoyannis IN.Recent Advances in Analytical Techniques used for the Determination of Fluoroquinolones in Pharmaceuticals and Samples of Biological Origin[J].Current Pharmaceutical Analysis,2005,1(2):155 - 193.
[2]Laban-Djurdjevic A,Jelikic-Stankov M,Djurdjevi?P.Optimization and validation of the direct HPLC method for the determination of moxifloxacin in plasma[J].J Chromatogr B,2006,844(1):104 - 111.
[3]Schulte S,Ackermann T,Bertram N,et al.Determination of the newer quinolones levofloxacin and moxifloxacin in plasma by high-performance liquid chromatography with fluorescence detection[J].J Chromatogr Sci,2006,44(4):205 -258.
[4]Nguyen HA,Grellet J,Ba BB,et al.Simultaneous determination of levofloxacin,gatifloxacin and moxifloxacin in serum by liquid chromatography with column switching[J].J Chromatogr B,2004,810(1):77 - 83.
[5]付丽娟,陈 蓉,涂晓真.几种氟喹诺酮药物含量测定的色谱条件比较及用HPLC法测定依诺沙星制剂的含量[J].中国药品标准,2005,6(1):17-18.
[6]陈 漪,施家威,金米聪.液相色谱-离子阱质谱法测定人血清中莫西沙星浓度[J].中国临床药学杂志,2009,18(6):345-348.
[7]李 红,陈光龙,殷静远,等.高效液相色谱法测定司帕沙星注射液的含量和有关物质[J].中国药业,2006,15(6):14 -15.
[8]孙曼春,方世平,王智勇,等.盐酸左氧氟沙星在健康人体内的药代动力学研究[J].中国药业,2010,19(3):5 -6.
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
世界最新医学信息文摘(2021年12期)2021-06-09
中成药(2018年6期)2018-07-11
中国粮油学报(2016年5期)2016-01-23
中国卫生标准管理(2015年25期)2016-01-14
中国当代医药(2015年36期)2015-03-11
中国药业(2014年21期)2014-05-26
郑州大学学报(理学版)(2014年2期)2014-03-01
中国信息化·学术版(2013年3期)2013-06-25
中国合理用药探索(2010年8期)2010-02-09
