
高效液相色谱法分析胞内S-腺苷-L-甲硫氨酸含量及其合成酶活性的优化研究
2011-07-25 12:36:10姚高峰秦秀林钱江潮
化学与生物工程 2011年6期
姚高峰,秦秀林,储 炬,钱江潮
(华东理工大学 生物反应器工程国家重点实验室,上海 200237)
S-腺苷-L-甲硫氨酸(S-Adenosyl-L-methionine,简称SAM)是一种重要的中间代谢物,广泛存在于微生物、动物、植物体内,可作为甲基、亚甲基、氨基、核糖、氨丙基的供体,参与体内的各种生化反应[1~3]。作为一种具有重要生理活性的物质,SAM在治疗肝炎、肝功能紊乱、关节炎、抑郁症、心血管等疾病以及抗衰老、防癌[4]等方面发挥着重要作用[5,6],市场需求日益增大。
生物反应器工程国家重点实验室通过在重组毕赤酵母(Pichiapastoris)中高效表达SAM合成酶,获得了能大量积累SAM的重组菌G/DS16[7]。但利用HPLC方法定量分析胞内SAM含量及SAM合成酶活性时存在保留时间长或峰宽、分离效果不佳的问题[8~10]。
针对胞内SAM抽提液及SAM合成酶测定体系中SAM的分离,作者对HPLC方法的流动相和洗脱程序进行优化,以建立一种分离效果好、精确度高、重复性好、保留时间适中的SAM定量分析方法,用于测定胞内SAM含量与SAM合成酶活性。
1 实验
1.1 材料、试剂与仪器
SAM标准品,Sigma公司;ATP、L-Met,上海生工生物工程有限公司;高氯酸、甲酸铵、甲酸均为分析纯;水为双蒸水。
Agilent HP1100型高效液相色谱仪,Thermo Scientific型冷冻离心机,QT-1型漩涡混合器,电子天平,恒温水浴锅,0.22 μm水相滤头等。
1.2 菌种
菌种G/DS16,自行构建保藏[7]。G/DS16为基因工程改造菌,甲醇诱导并补加甲硫氨酸后胞内SAM合成酶活性增大,SAM含量较高。
1.3 色谱条件
Thermo-BioBasic SCX色谱柱(4.6 mm×250 mm,5 μm),流动相为甲酸铵溶液(pH值4.0),流速1 mL·min-1,检测波长254 nm,柱温25 ℃,进样量10 μL。
1.4 流动相的配制
准确称取31.5 g甲酸铵溶于水中,定容至1000 mL,配制成浓度为0.5 mol·L-1的甲酸铵溶液,用甲酸调pH值4.0左右。取0.5 mol·L-1甲酸铵溶液适量,稀释至终浓度为5 mmol·L-1,用甲酸调pH值4.0左右。流动相现配现用,0.22 μm水相滤头过滤。
1.5 标准溶液的配制
准确称取SAM标准品25 mg,加水溶解,定容,配制成浓度为1.0 g·L-1的标准品储备液。取适量标准品储备液加水稀释,得浓度分别为0.5 g·L-1、0.35 g·L-1、0.25 g·L-1、0.15 g·L-1、0.05 g·L-1的标准溶液。进样前用0.22 μm水相滤头过滤。
1.6 样品制备
1.6.1 胞内SAM抽提样品
取G/DS16发酵液1 mL,12 000 r·min-1离心2 min,去上清,加入1 mL10%三氯乙酸(TCA)重悬,于4 ℃静置抽提2 h,离心后取上清液,用0.22 μm水相滤头过滤,待测。
1.6.2 SAM合成酶活性测定样品
发酵过程中SAM合成酶的酶活水平是通过测定体外酶促反应液的SAM含量来监控的。取1 mL G/DS16发酵液,离心去上清,加1 mL缓冲溶液和等体积的酸洗玻璃珠,按冰水浴30 s、振荡破碎30 s交替进行8次,12 000 r·min-1、4 ℃离心7 min以上,上清即为粗酶液。SAM合成酶活性测定反应体系见表1。

表1 SAM合成酶活性测定反应体系
反应体系在37 ℃反应1 h,加500 μL 20%高氯酸,4 ℃放置0.5 h以上终止反应,12 000 r·min-1、4 ℃离心10 min以上,取上清,用0.22 μm水相滤头过滤,待测。以不加粗酶液、ATP或L-Met的反应体系作为对照。
2 结果与讨论
2.1 流动相与洗脱程序的优化
首先以普遍使用的0.5 mol·L-1(pH值4.0)甲酸铵作为流动相进行HPLC测定,结果见图1。

★—SAM —杂质(a)SAM标准品 (b)SAM合成酶活性测定对照组 (c)SAM合成酶活性测定样品 (d)胞内SAM抽提样品
由图1可知,对于SAM合成酶活性测定反应体系,0.5 mol·L-1甲酸铵溶液作为流动相的分离效果不佳。SAM合成酶活性测定对照组分离时在SAM峰相应处出现一个杂质峰(图1b),这可能是由于其中含有ATP、L-Met以及反应生成的一些杂质,这些物质的保留时间与SAM相同,造成了对SAM测定的干扰。而检测SAM合成酶活性测定样品时,杂质峰与SAM峰重叠(图1c),严重影响了测定结果的准确性。而且,采用0.5 mol·L-1甲酸铵为流动相,5 min之内可完成洗脱过程,SAM保留时间过短,即使在胞内抽提样品中,也存在保留时间相近的其它物质(图1d),严重影响了分离效果。
为了获得较好的分离效果,针对流动相和洗脱程序进行优化。通过改变流动相中的甲酸铵浓度来调节保留时间,优化的洗脱程序如下:以5 mmol·L-1的甲酸铵洗脱5 min,去除大量杂质;然后提高甲酸铵的浓度(0.5 mol·L-1甲酸铵∶5 mmol·L-1甲酸铵=9∶1)洗脱4 min;再用0.5 mol·L-1的甲酸铵洗脱3 min,将SAM洗脱;最后用5 mmol·L-1的甲酸铵重新平衡柱子。整个洗脱过程中,流动相的pH值为4.0,流速为1 mL·min-1。
在优化的流动相和洗脱程序下进行HPLC测定,结果见图2。

★—SAM —杂质(a)SAM标准品 (b)SAM合成酶活性测定对照组 (c)SAM合成酶活性测定样品 (d)胞内SAM抽提样品
由图2可知,SAM标准品保留时间为10.1 min左右,峰形较好(图2a);SAM合成酶活性测定对照组样品中的杂质与SAM得到了很好的分离(图2b);SAM合成酶活性测定样品以及胞内SAM抽提样品中SAM组分都得到了有效分离,峰宽、峰形较理想,没有杂质峰的干扰(图2c、d)。这说明适合的SAM保留时间排除了保留时间短可能造成的溶剂峰干扰,且相对于反相C18柱法较长的保留时间[11],该方法的检测效率明显提高。
2.2 SAM的标准曲线
取浓度为0.05~0.5 g·L-1的SAM标准溶液,采用优化的流动相和洗脱程序进行HPLC分析,以峰面积(y)对SAM浓度(x)绘制标准曲线,见图3。
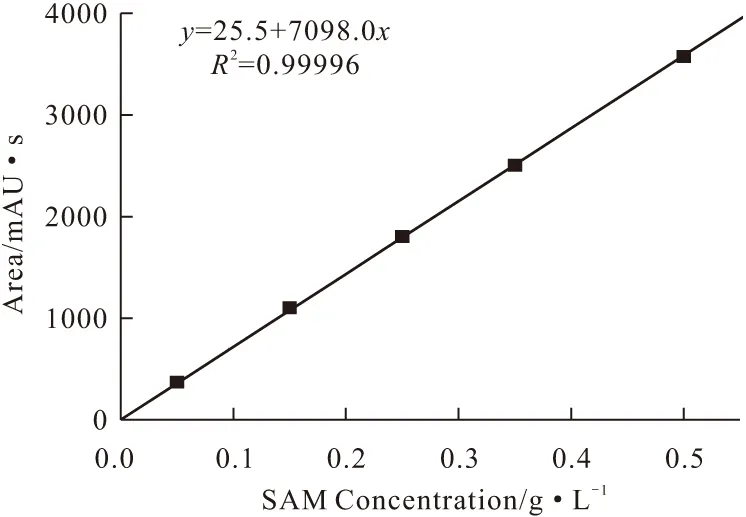
图3 SAM的标准曲线
由图3可知,在0.05~0.5 g·L-1浓度范围内,SAM的浓度和峰面积线性关系良好,标准曲线方程为y=25.5+7098.0x,R2为0.99996。
2.3 精密度
取10 μL 250 mg·L-1的SAM标准溶液进样,平行测定5次,结果见表2。
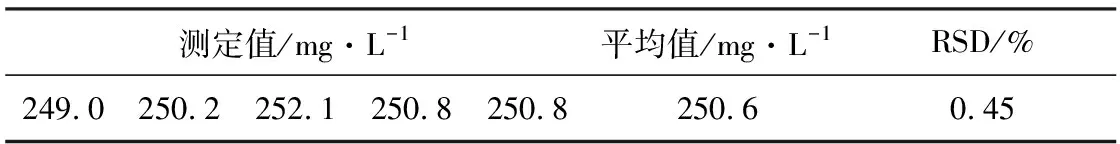
表2 精密度实验结果(n=5)
由表2可知,相对标准偏差为0.45%,表明该方法精密度良好。
2.4 重复性
分别取同一SAM合成酶活性测定样品和胞内SAM抽提样品10 μL进样,平行测定5次,结果见表3。

表3 重复性实验结果(n=5)
由表3可知,相对标准偏差分别为0.49%和0.59%,表明该方法重复性良好。
2.5 回收率
分别取已知浓度SAM合成酶活性测定样品和胞内SAM抽提样品各6份,3份一组,各添加2个不同浓度(100 mg·L-1、300 mg·L-1)的SAM标准品,按优化色谱条件进样,结果见表4。

表4 加样回收率实验结果
由表4可知,平均回收率为99.3%~100.5%,相对标准偏差为0.72%~0.98%,表明该方法回收率高,准确可靠。
2.6 稳定性
取同一SAM合成酶活性测定样品和胞内SAM抽提样品,每隔2 h进样测定,平行测定5次,结果见表5。

表5 稳定性实验结果(n=5)
由表5可知,相对标准偏差分别为0.76%和0.69%,表明该方法稳定性良好,能够适用于长时间、多样品的测定。
2.7 SAM合成酶活性测定对照组的考察
在SAM合成酶活性测定过程中需要设置对照组以扣除本底水平的SAM,一般以不加ATP(对照组1)或不加L-Met(对照组2)为对照组。针对两种情况进行考察,结果见表6。
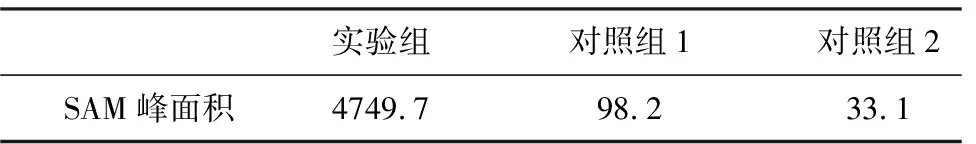
表6 SAM合成酶活性测定对照组的考察
由表6可知,实验组的SAM峰面积为4749.7,而对照组1为98.2、对照组2为33.1,两对照组SAM峰面积差别较小,仅占实验组测定值的1%左右,对测定结果的干扰有限。粗酶液中含有少量的胞内水平的ATP和L-Met,ATP作为一种活性物质,含量较低且稳定性较差,而L-Met相比而言较为稳定。推测用于酶活测定的粗酶液中残留的L-Met参与了对照组1的反应,直接导致了两对照组SAM含量的差异,因此,选择不加ATP的对照组更能反映实际的酶活水平。
3 结论
分析了原有HPLC方法测定SAM存在的问题,针对流动相和洗脱程序进行优化,建立了一种高效、稳定、准确性高的SAM定量检测方法。确定最佳HPLC条件为:254 nm吸收波长下,以5 mmol·L-1和0.5 mol·L-1的甲酸铵在不同时间段以1 mL·min-1的流速进行洗脱。在此色谱条件下,胞内SAM抽提样品和SAM合成酶酶促反应样品中的SAM都能够得到有效分离。该方法简单高效、重复性好、精确度高,并且性能稳定,可以较长时间进行大量样品的准确测定,能够满足发酵过程中菌体胞内SAM产量的测定以及SAM合成酶酶活的监控要求。
[1] Mato J M,Alvarez L,Ortiz P,et al.S-Adenosylmethionine synthesis:Molecular mechanisms and clinical implications[J].Pharmacology & Therapeutics,1997,73(3):265-280.
[2] 项昱红,莫晓燕,詹谷宇.S-腺苷甲硫氨酸的制备与药理作用[J].西北药学杂志,1999,14(1):38-39.
[3] 杨静,王旻,韦平和.S-腺苷甲硫氨酸的临床及药理研究进展[J].药学进展,2001,25(3):164-167.
[4] Qiu W,Zhou B,Chu P G,et al.The induction of growth arrest DNA damage-inducible gene 45 beta in human hepatoma cell lines byS-adenosylmethionine[J].American Journal of Surgical Pathology,2007,171(1):287-296.
[5] Bottiglieri T.S-Adenosyl-L-methionine (SAMe):From the bench to the bedside——molecular basis of a pleiotrophic molecule[J].American Journal of Clinical Nutrition,2002,76(5):1151S-1157S.
[6] Bailey S M,Robinson G,Pinner A,et al.S-Adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver[J].American Journal of Physiology-Gastrointestinal and Liver Physiology,2006,291(5):857-867.
[7] Hu H,Qian J C,Chu J,et al.DNA Shuffling of methionine adenosyltransferase gene leads to improvedS-adenosyl-L-methionine production inPichiapastoris[J].Journal of Biotechnology,2009,141(3-4):97-103.
[8] 胡辉.S-腺苷甲硫氨酸高产菌株的构建及其发酵合成SAM优化策略[D].上海:华东理工大学,2008.
[9] 张建国,王学东,魏东芝.高效液相色谱法同时测定Pichiapastoris发酵过程中腺苷甲硫氨酸相关的六种物质[J].工业微生物,2008,38(1):6-9.
[10] 蔡恒,郑伟刚,万红贵,等.HPLC-UV法定量测定发酵液中的S-腺苷-L-甲硫氨酸[J].中国生物工程杂志,2007,27(11):73-76.
[11] Wang W,Kramer P M,Yang S,et al.Reversed-phase high-performance liquid chromatography procedure for the simultaneous determination ofS-adenosyl-L-methionine andS-adenosyl-L-homocysteine in mouse liver and the effect of methionine on their concentrations[J].Journal of Chromatography B:Biomedical Sciences and Applications,2001,762(1):59-65.
猜你喜欢
食品科学(2023年13期)2023-08-12 00:41:48
中国生物化学与分子生物学报(2022年7期)2022-09-07 05:28:48
检察风云(2022年5期)2022-04-05 13:42:39
中国药科大学学报(2021年6期)2021-12-31 03:05:20
传染病信息(2021年6期)2021-02-12 01:52:08
中成药(2017年12期)2018-01-19 02:06:27
福建轻纺(2017年12期)2017-04-10 12:56:39
国外医药(抗生素分册)(2016年3期)2016-07-12 14:25:18
山东医药(2015年16期)2016-01-12 00:40:08
分析测试学报(2015年9期)2015-12-17 16:44:28
