
草酸热分解机理的DFT研究
2011-07-25 03:48:02王冬梅耿志远
化学与生物工程 2011年3期
王冬梅,耿志远
(1.宝鸡文理学院化学化工系,陕西 宝鸡 721013;2.西北师范大学化学化工学院,甘肃 兰州 730070)
草酸是脂肪族二元羧酸中分子量最小的酸,关于草酸的性质,已有广泛的实验及理论研究。研究表明,不论光照或是加热,草酸均可以分解生成CO、CO2、H2O、HCOOH等物质。
有关草酸光解的研究,早在1985年,Yamamoto等[1]对草酸在气相中第一激发态下的光解离进行了研究,温度在115℃时,得到的产物为CO、CO2、H2O和HCOOH。1992年,Nieminen等[2]又在Ne、Ar和Xe气体模型中,对草酸的解离作了光谱学分析,也得到同样的产物。相应地,有关草酸的热分解,文献记载:温度在453 K时,草酸分解产物仅有CO2和HCOOH,在多光子红外作用下,CO和H2O才是最重要的产物。虽已有大量的实验事实,而从理论上用速率常数解释最优解离途径的报道并不多见。因此,作者根据前人实验的结果,对草酸的热分解提出了三条可能的解离途径:
(1)H2C2O4→HCOOH+CO2
(2)H2C2O4→(HO)2C+CO2
(HO)2C→H2O+CO
(3)H2C2O4→CO2+CO+H2O
通过计算每条反应途径的活化能及速率常数,选出了最优解离通道。得到了与实验[1]一致的结果。
1 计算方法
采用密度泛函理论中的B3LYP方法,用6-311++G**基组,对三种途径中的反应物、中间体、过渡态及产物进行了几何构型优化,优化得到的构型见图1~图3。对中间体和过渡态的频率分析表明:所有中间体的力常数本征值为正,说明它们是反应势能面上的稳定点,各过渡态均有唯一的虚频(见表1),随后对其进行的IRC计算确认了这些过渡态的正确性。最后用CCSD(T)/6-31G**方法在B3LYP/6-311G**构型下计算了各驻点的能量,以确定能级顺序,所有的能量均进行了零点能校正。根据经典过渡态理论,反应绝对速率常数k可由式(1)确定:
k=λ(kBT/h)(Q#/QAQB)exp(-Ea/RT)
(1)
式中:λ为考虑到量子效应引进的校正因子,通常λ=1;Q#、QA(或QB)分别为过渡态、反应物的配分函数;Ea为反应的活化能。配分函数Q为体系平动、转动和振动配分函数的积,即Q=QtQrQv,所有计算均用Gaussian03完成。
2 结果与讨论
2.1 H2C2O4热分解机理的研究
2.1.1 H2C2O4分解为HCOOH和CO2的反应
在B3LYP/6-311G**的理论水平上,对反应通道1的各驻点进行了几何优化,几何参数见图1,能量见表1。由草酸最稳定的反式构型R开始,H8经过一个四中心的过渡态TS1向C1迁移,对过渡态TS1进行了振动频率分析,结果仅有一个虚频,其中虚频2074.02icm-1对应的振动模式为C1-C2和H8-O6的伸缩振动,如图1中的TS1构型。在此过程中,C1与C2、O6与H8的距离逐渐伸长,并且断裂,C1和H8成键,生成产物P1(HCOOH+CO2)。
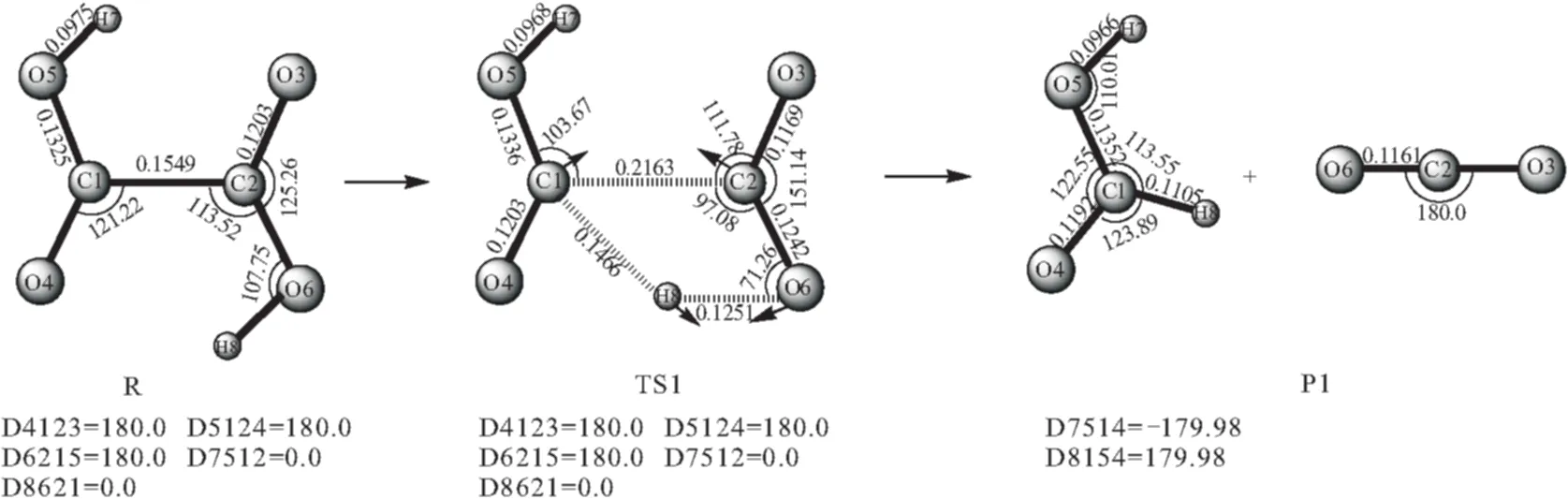
图1 H2C2O4分解为HCOOH和CO2的路径示意图
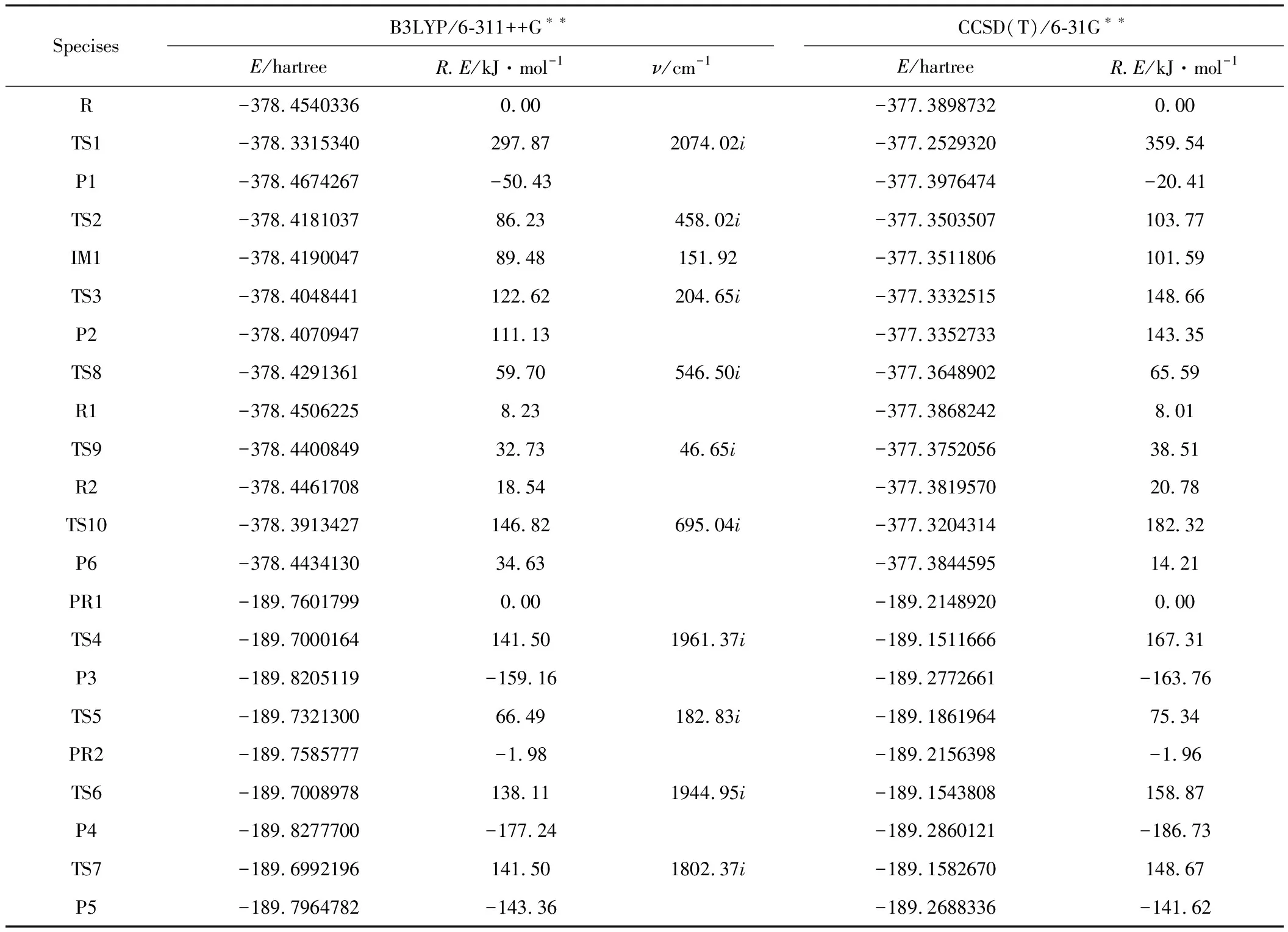
表1 反应物、中间体、过渡态及产物能量(a.u)以及过渡态的振动频率
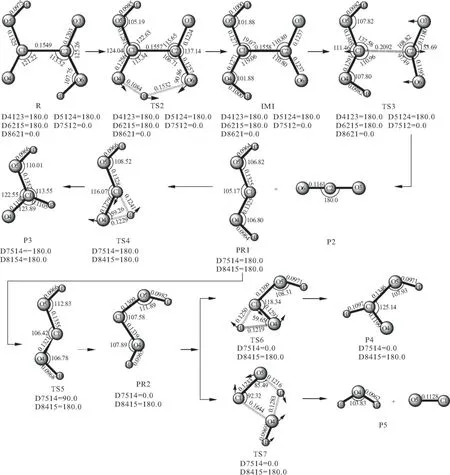
图2 H2C2O4分解为HCOOH、CO、CO2、H2O的路径示意图
从图1可看出,这是一个一步完成的基元反应,H2C2O4中C1-C2键长为154.85 pm,长于正常的C-C单键,但加热下,C-C键是非解离态,其它的C=O、C-O和O-H键都在正常键长的范围内,也与文献[3]的计算结果一致。在此过程中由R到TS1要跨越297.87 kJ·mol-1的能量,解离比较困难。其能量关系如图4所示。
2.1.2 H2C2O4分解为HCOOH、CO、CO2、H2O的反应
由反应物R开始,H8经过一个五元环的过渡态TS2向O4上迁移,对过渡态TS2进行振动分析,结果仅有一个虚频,其中虚频458.02icm-1对应的振动模式为O4-H8-O6的伸缩振动,如图2中的TS2构型。此过程中H8与O6的距离伸长并断裂,O4与H8距离缩短并成键,形成了中间体IM1,该步反应只需跨越86.23 kJ·mol-1的能垒。随着中间体IM1中C1和C2键的伸长,经过渡态TS3,同样对过渡态TS3进行振动分析,结果仅有一个虚频,其中虚频204.65icm-1对应的振动模式为C1-C2的伸缩振动,如图2中的TS3构型。该过程的能量关系见图4,可以看出很容易跨越33.14 kJ·mol-1的能垒,生成产物P2(HCOOH:+CO2)。产物P2中的PR1(HCOOH:)虽为稳定构型,但能量较高,易形成异构体或分解。PR1(cis-HCOOH:)经TS4,O4与H8键逐渐伸长并断裂,同时,C1与H8成键,该步反应需跨越141.50 kJ·mol-1的能垒形成产物P3(cis-HCOOH)。对过渡态TS4进行振动分析,结果仅有一个虚频,其中虚频1961.37icm-1对应的振动模式为C1-O4-H8的伸缩振动,如图2中的TS4构型。另外,PR1(cis-HCOOH:)经内转换过渡态TS5形成稳定中间体PR2(tran-HCOOH),该步仅需66.49 kJ·mol-1的能量 。再由中间体PR2开始,经TS6,H8与O4断键,与C1成键,跨越151.44 kJ·mol-1的能垒生成P4(cis-HCOOH)。对过渡态TS6进行振动分析,结果仅有一个虚频,其中虚频1944.95icm-1对应的振动模式为C1-H8-O4的伸缩振动,如图2中的TS6构型。此外,中间体PR2还可经过TS7形成一个四元环,进行分子内脱水得到产物P5(CO+H2O),对过渡态TS7进行振动分析,结果仅有一个虚频,其中虚频1802.37icm-1对应的振动模式为C1- O5和H7-O4-H8的伸缩振动,如图2中的TS7构型,能量关系见图4。该过程只需143.48 kJ·mol-1的能量。从该途径整体上来看,无论形成中间体或生成产物,所需要跨越的能垒均不高。可见,此过程为反应物R解离的优选方式。
2.1.3 H2C2O4分解为H2O、CO、CO2的反应
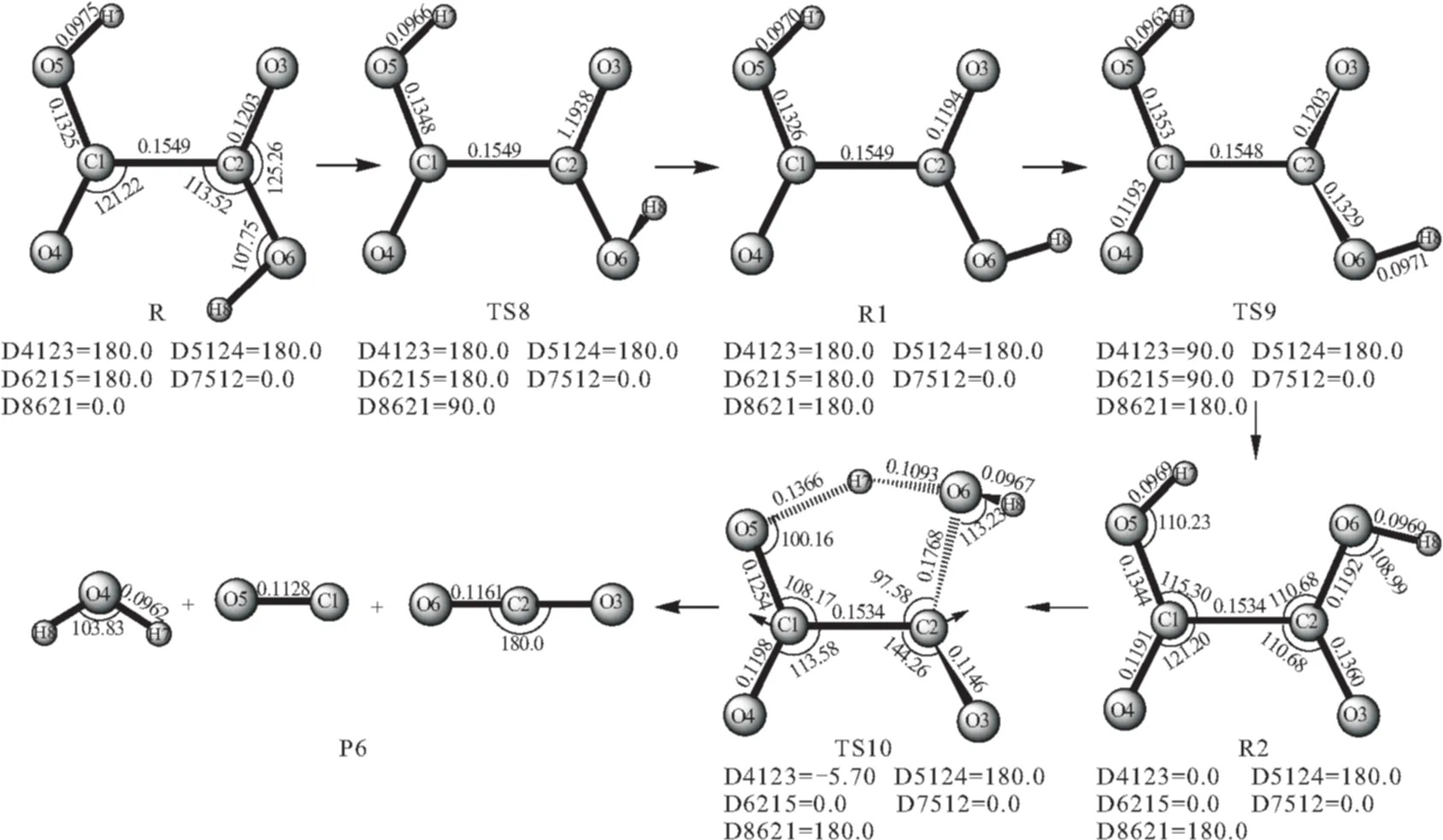
图3 H2C2O4分解为H2O、CO、CO2的路径示意图
从构型来看,直接由反应物R进行分子内脱水似乎不大可能,然而其异构体R2(cis-HOOCCOOH)经实验检测,可以进行分子内脱水反应,由R→R2可经过TS8→R1→TS9,其中TS8和TS9分别为内转化过渡态,经频率分析,有唯一的虚频,见表1。从能量的角度讲,由R→R2仅相差18.54 kJ·mol-1,与文献[4,5]相符。
因此,该途径由反应物R2开始,经一个扭转构型的过渡态TS10,对该过渡态进行振动分析,结果仅有一个虚频,其中虚频695.04icm-1对应的振动模式为C1-C2的伸缩振动,如图3中的TS10构型,能量关系见图4。此过程中,C2与O6距离伸长并断键;O5与H7的距离也伸长,H7成功迁移到O6上,生成了H2O;C1与C2键也解离开,生成了CO和CO2。整个过程中,由TS10到P6(H2O+CO+CO2)的生成,只需跨越128.28 kJ·mol-1的能垒,且得到产物的顺序几乎是同时的。可见,该过程也是一协同反应,反应物R2的解离比较容易。其能量关系见图4。
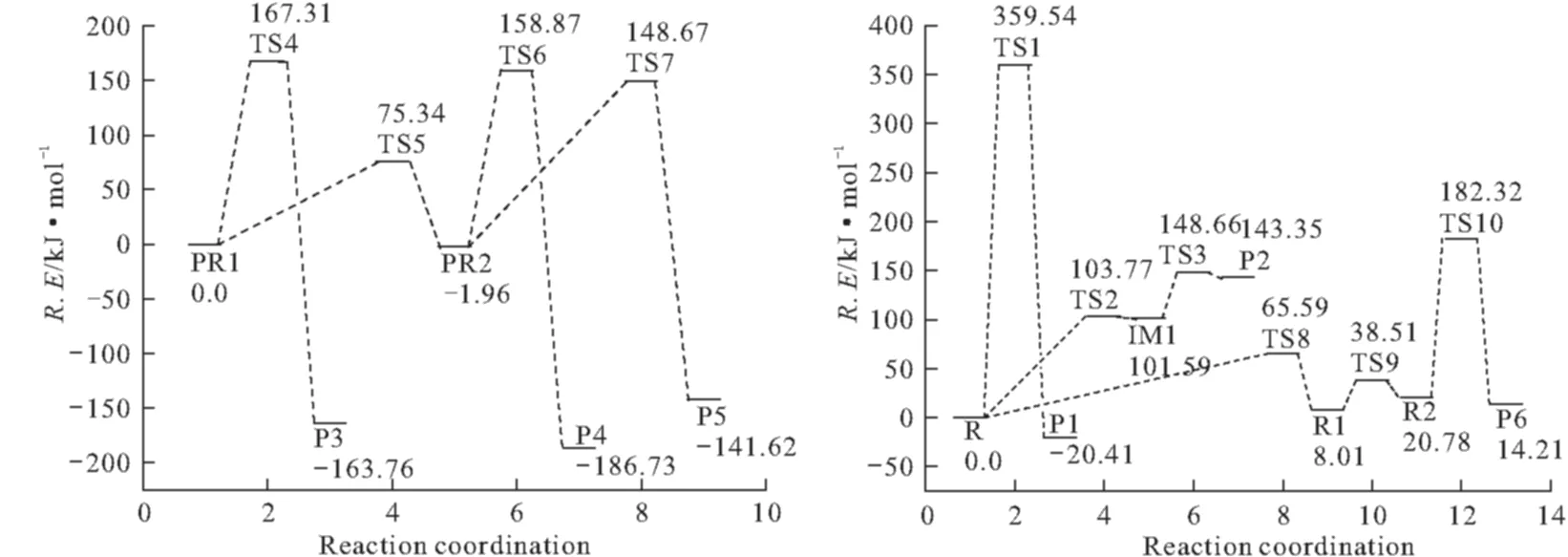
图4 H2C2O4解离的势能面示意图
2.2 反应速率常数的理论计算
为了解温度对草酸热解反应路径的影响,根据经典过渡态理论反应速率常数公式(1)计算了298~800 K范围内草酸三种热解离路径的速率常数k,见表2。第一条解离通道:R-TS1-P1过程反应活化能较大,将过渡态TS1及反应物R计算得到的配分函数代入公式(1),发现在两种水平下得到的k值均很小,直到温度上升至800 K时,草酸才有分解的可能性。第二条解离通道,经由R-TS2-IM1-TS3-P2而完成,且控速步骤为R-TS2。由于该步为连续反应,故计算了总反应速率常数,得到了与400 K时实验结果很吻合的值。第三条解离通道,经由R-TS8-R1-TS9-R2-TS10-P6而完成,其中R-R2为异构体转化,仅需18.54 kJ·mol-1的能量。因此只将R2-TS10-P6过程的活化能及相应的配分函数代入公式(1)进行了计算。由表2数据可知,随着温度的升高,反应速率加快。在温度为400~800 K范围内,途径2和途径3热解均可以发生。

表2 不同温度下各热解路径的速率常数k
3 结论
采用量子化学中的DFT方法,在B3LYP/6-311++G**基组水平上对草酸的热分解机理进行了研究,计算了298~800 K范围内草酸三种热解离路径的速率常数。结果表明,H2C2O4热分解有两条可行的反应途径:R-TS2-IM1-TS3-P2和R-TS8-R1-TS9-R2-TS10-P6。
[1]Yamamoto S,Back R A.The gas-phase photochemistry of oxalic acid[J].J Phys Chem,1985,89(4):622-625.
[2]Nieminen J,Rasanen M,Murto J.Matrix-isolation and ab initio studies of oxalic acid[J].J Phys Chem,1992,96(13):5303-5308.
[3]Van Alsenoy C,Klimkowsk V J,Schäfter L.Ab initio studies of structural features not easily amenable to experiment.Part 37.Structural and conformational investigations of the dicarbonyls gloxal,biacetyl and oxalic acid[J].J Mol Struct:THEOCHEM,1984,109(3-4):321-330.
[4]Nahlovska Z,Nahlovsky B,Strand T G.Molecular structure of gaseous oxalic acid from electron diffraction and IR data[J].Acta Chem Scand,1970,24(7):2617-2628.
[5]Ajo D, Condorelli G,Fragala I,et al.Conformation and electronic structure of oxalic acid by the ab initio method[J].J Mol Struct,1977,37(1):160-163.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
大学化学(2021年8期)2021-09-26 10:51:16
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
电脑知识与技术(2018年3期)2018-03-21 09:27:04
中学化学(2017年5期)2017-07-07 08:40:47
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
中学化学(2016年4期)2016-05-30 16:20:37
中学化学(2014年1期)2014-04-23 08:59:04
