
碳纳米管负载的CoB和CoBP对磷化氢的分解性能
2011-07-19 06:37李俐俐周逸潇韩新宇张宝贵
天津大学学报(自然科学与工程技术版) 2011年11期
李俐俐,杨 丽,周逸潇,韩新宇,张宝贵
(1. 南开大学环境科学与工程学院,天津 300071;2. 周口师范学院生命科学系,周口 466000)
磷化氢(PH3)为易燃的高毒气类体.一定量的磷化氢通过呼吸道进入人和动物体内后,将引起以神经系统、呼吸系统损害为主的全身性疾病.PH3气体排放到大气中后,因为该种气体具有强还原性,会消耗大气中的自由基,所以具有间接的温室效应[1].工业上,多种磷化工产品的生产、半导体器件的制备以及乙炔制造等过程中,都会产生 PH3尾气.目前,对PH3尾气的处理方法主要有:①利用液态和固态氧化剂还原 PH3的还原法;②采用吸附剂对 PH3进行物理和化学吸附的吸附法;③在燃烧炉内将PH3与空气混合燃烧的燃烧法[2].但这些方法是将PH3最终转化为磷的氧化物、水溶性酸或盐类,在减少二次污染、提高经济效益等方面还有待进一步提高.20世纪 90年代后,由于污染严重,国际上的磷化工企业大量迁移至不发达国家,我国已成为世界上磷化工产品的生产大国.因此,深入探索对环境友好、工业附加值高的PH3处理技术具有十分重要的社会意义.
将 PH3催化分解为磷(P)和氢气(H2)的思路在20世纪 80年代首次被提出[3],近年内已在多种铁系金属与类金属(硼、磷)组成的合金(如 FeCuP[4]和非晶态 CoNiBP/CNTs[4]、CoB[5]等)上证实采用该类方法处理 PH3尾气具有长远而现实的环境和经济效益.一方面,磷和氢气的经济价值较大;另一方面,适宜的催化剂和反应温度可使 PH3尾气达到接近完全分解.其中,CoNiBP/CNTs非晶态合金表现出了最高的分解活性.CoNiBP/CNTs在 380,℃、PH3空速为2,520,mL/(h·gcat)时,PH3分解率可达 99.7%[4].研究将CoNiBP/CNTs的高活性归因于合金颗粒的高度分散及碳纳米管(carbon nanotubes,CNTs)作为催化剂的载体所拥有的优异性能.但总体而言,前期对 PH3催化分解的报道主要集中于有关催化剂的制备和催化性能方面的研究,对PH3催化分解的反应机制以及合金中P和(或)B组分对催化性能的影响涉及很少.
笔者以 CNTs为载体,用诱导-化学还原法制备了非晶态 CoB/CNTs和 CoBP/CNTs,用于 PH3催化分解.考察了催化剂中类金属 B、P含量的变化对催化性能的影响,并对由 CoB/CNTs和 CoBP/CNTs产生的催化剂进行了一系列表征,结合反应机制探讨了PH3催化分解中过程CoB/CNTs和CoBP/CNTs活性相的差异,以期进一步深化PH3催化分解催化剂的研究和开发.
1 实验方法
1.1 催化剂的制备和处理
1.1.1 CNTs的预处理
将多壁碳纳米管(直径为 10~20,nm,纯度>95%,购自中国深圳纳米技术有限公司)浸渍于体积比为1∶1的浓H2SO4和浓HNO3混合液中,在120,℃回流 6,h,进行氧化处理.再依次放入浓氨水,在60,℃回流 2,h.最后放入 0.25,mol/L柠檬酸水溶液中,溶液沸点回流 2,h,对 CNTs进行修饰.所得CNTs经过滤、洗涤和干燥后,备用.
1.1.2 非晶态CoB/CNTs和CoBP/CNTs催化剂的制备
采用诱导-化学还原法.首先,将预处理的 CNTs浸渍于 CoCl2·6H2O 水溶液中,滴加几滴 KBH4溶液,使CNTs负载上少量的CoB微晶核.一系列不同P与(B+P)摩尔比的 CoBP/CNTs制备过程如下:取适量负载少量 CoB微晶核的 CNTs,置于溶有Na3C6H5O7·2H2O、(NH4)2SO4、CoCl2·6H2O 和NaH2PO2·H2O的水溶液中,加入不同体积的2.0,mol/L KBH4溶液还原.反应在室温下进行,并在N2气流保护下持续搅拌,直至无气泡产生.CoB/CNTs的制备过程与 CoBP/CNTs的制备类似,但反应液中不含 NaH2PO2·H2O.所制得的黑色沉积物经充分洗涤后,用 N2(含体积分数为 1%的 O2)钝化(记为制备的催化剂).
1.1.3 CoB/CNTs和CoBP/CNTs样品的煅烧处理
分别在 300,℃和 500,℃,H2气流中进行,处理时间为2,h,温度降至室温后,钝化,进行XRD检测.
1.2 催化剂的表征
体相组成用(ICP-9000(N+M)电感耦合等离子体发射光谱仪测定.结构用 Rigaku-D/MAX 2500型X-射线衍射(XRD)仪进行测定,用 CuKα为辐射源(λ=0.154 18 nm).比表面积(SBET)采用 77,K下N2吸附法测量,测定在 Quantachrome Autosorb-1吸附仪中进行.形貌采用 JEM-2100高倍透射电子显微镜(HRTEM)进行观察.表面组分和电子状态测定在Kratos Axis Ultra DLD multi-technique X-射线光电子能谱(XPS)仪中进行.
1.3 活性测定
PH3分解反应在常压固定床反应器中进行.反应前,用连续流动的 H2气流在 200,℃下还原 2,h.以PH3和N2(含体积分数为5%的PH3)混合气为反应气体.催化活性测试的条件为:300~460,℃,常压,PH3气体空速 2,520,mL/(h·gcat).通过在线气相色谱火焰光度检测器(FPD)进行产物分析,配置的色谱柱为Poropak Q型.活性测试后,样品在流动的N2下冷却至室温,钝化(称为使用过的催化剂),对之进行一系列检测.
2 结果与分析
2.1 TEM的表征
图 1为制备的CoB/CNTs和CoBP/CNTs(P与(B+P)的摩尔比 n=0.43)高倍透射电镜照片,从该图中可见,球形的 CoB、CoBP颗粒均匀地分散于CNTs载体上.CoB颗粒直径平均约为 15,nm,CoBP颗粒直径略大,约为 18,nm.两者颗粒大小的差异可归因为BH4-的还原能力远强于 H2PO2-,使 Co2+快速还原,因而阻碍了 CoB颗粒的增长.另外,从图中可见球形颗粒外周包覆着一层浅灰色氧化层薄膜.钝化处理可使纳米颗粒表面形成一层氧化薄膜,保护颗粒内部,避免发生深度氧化.
2.2 XRD分析
图2 显示了制备的CoB/CNTs(曲线1)和CoBP/CNTs(曲线2,n=0.43)以及2个样品用H2气流在不同温度下煅烧2,h的XRD图谱(曲线3~6).图2曲线1和2中,除了在2θ=26°出现了CNTs的石墨碳衍射峰外,未见其他晶相峰,仅在 2θ=45°附近出现了典型的非晶态结构所具有的宽泛弥散峰[6],说明制备的样品均为非晶态结构.由于非晶态结构属于热力学的亚稳态,因而在外界条件(如高温)的影响下会发生相分离、晶化,导致结构发生变化.对样品热处理结果证实了这一特点.图2中曲线3和4为300,℃下 H2气流处理上述非晶态样品的 XRD图谱,由图可见,2个样品中均出现了较弱的金属Co的衍射峰,说明2种非晶态样品在该温度下已经发生晶化.当加热温度提高到 500,℃时,CoB/CNTs(曲线 5)样品中出现了尖锐金属Co衍射峰,而CoBP/CNTs(曲线6)样品中除了出现了尖锐金属 Co衍射峰外,还出现了Co2P的晶态峰,说明非晶态结构在此温度下进一步晶化,并伴随着 CoB完全分解为晶态的 Co以及CoBP完全分解为晶态的Co和Co2P.在2种非晶态样品晶化过程中,未见金属硼化物的晶化相出现,说明B均匀地分散于载体CNTs上.
前期关于CoNiBP/CNTs催化PH3分解的研究表明,在PH3催化分解过程中,产物P有极少量以原子形式迁移进合金颗粒,与负载合金中的金属很快形成金属磷化物(CoP),所形成的磷化物 CoP较非晶态CoNiBP具有更高的催化活性和稳定性[4].制备的CoB/CNTs和 CoBP/CNTs(n=0.43)样品经过 300~440,℃温度范围内反应 8,h后,将使用过的样品进行检测,所得 XRD图谱如图 3所示.在2种使用过的催化剂中,均明显出现了CNTs和CoP化合物的衍射峰.另外,CoB/CNTs 样品中在 2θ=44.2°出现了一个尖锐的立方 Co衍射峰.通过对 CoB/CNTs和CoBP/CNTs样品反应前后的XRD检测结果可知,在PH3催化分解过程中,非晶态 CoBP/CNTs在高温下分解生成的晶态Co和Co2P全部被磷化成CoP.而对于CoB/CNTs,晶化产物Co只有一部分被产物磷化成CoP,还有一部分仍以金属 Co形式存在.XRD表征结果说明,大量B的存在对金属磷化具有抑制作用.
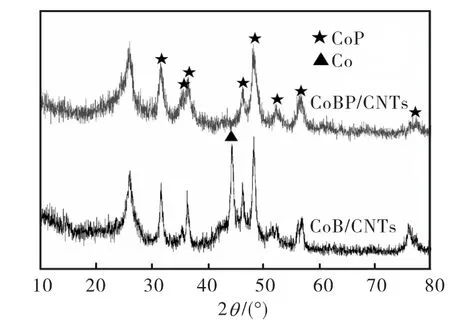
图3 使用过的催化剂的XRD谱Fig.3 XRD patterns of spent catalysts
2.3 反应前后的组成和比表面积
一系列不同P与(B+P)摩尔比的CNTs负载Co基非晶态合金样品的负载量,以及用于PH3分解反应前后的体相组成和比表面积列于表 1.由表 1可知,各样品的金属负载量基本相近.但反应前后,各样品的体相组成变化很大.反应后,各样品中 P的含量大增,这种变化趋势是由于载体CNTs上的Co与反应产物中极少量的P原子形成了CoP化合物.样品反应后各样品P和Co的摩尔比均大于1,说明有少量的P过量.另外,表1也显示反应后各样品的比表面积(SBET)均有所降低,可归因于以下 2个原因:①P原子迁入合金,形成 CoP化合物会使催化剂的密度有所增加;②反应过程中磷化物的形成和过量 P的存在导致催化剂孔的部分堵塞.
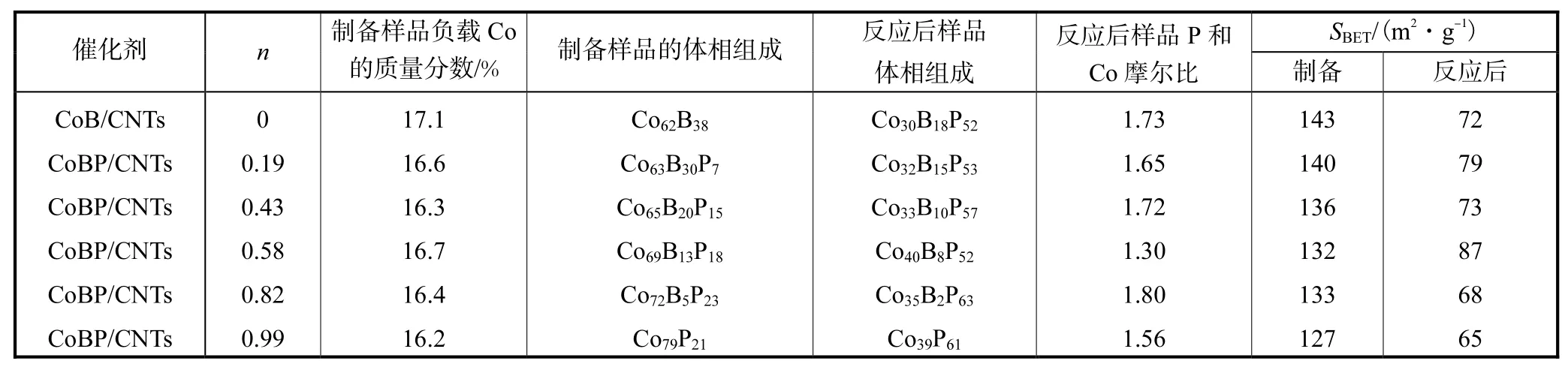
表1 CoB/CNTs和CoBP/CNTs反应前后的组分和比表面积Tal.1 Composition and surface area of as-prepared and spent CoB/CNTs and CoBP/CNTs
2.4 XPS检测
将使用过的CoB/CNTs和CoBP/CNTs(n=0.43)钝化处理后进行 XPS检测,检测结果如图 4所示.由 2个样品的 Co2p芯能级谱(见图 4(a))可以看出,芯能级谱均显示 2个峰.第 1个峰分别出现在 782.1,eV和 781.8,eV,该处吸收峰可归因于Co2+[7],推测是由 CoP表面氧化导致 Co以磷酸盐形式出现[8-9].第 2个峰集中于 779.1,eV 左右,根据XRD衍射结果,该峰对应于反应过程中形成的 CoP中 Co的束缚能,相比于金属 Co的标准的束缚能(778.4,eV),CoP中 Co的束缚能高于金属 Co,说明CoP中的Co带有部分正电荷.文献[8-9]指出CoP中Co的束缚能在779.9~778.6,eV之间.使用后催化剂中芯能级谱见图 4(b),在束缚能 134.4,eV附近和 129.7,eV附近可观察到 2个吸收峰.在 134.4,eV附近处的峰为典型的磷酸盐种类[10],是由磷化物表面氧化产生的.在 129.7,eV附近的吸收峰归因于CoP中的 P,与文献[8-9]报道值 128.8~129.7,eV 相似.CoP中P的束缚能较P标准束缚能(130,eV)低,说明P带有部分负电荷.电子从金属转移到P的现象已报道于多种金属磷化物[10-11].B1s芯能级谱(见图4,c)显示,使用过的样品中 B既以元素态(B)形式存在,也以氧化态(BO2-)形式存在,分别对应于束缚能187.6,eV、187.4,eV 和 192.2,eV、192.1,eV 处的吸收峰[12].样品中元素态 B的束缚能 187.6,eV、187.4,eV均高于B的标准束缚能(187.1,eV),说明2样品中的元素B带有部分正电荷.通过对使用过的样品的XPS检测,可推断出,反应过程中形成的CoP中Co带有部分正电荷,呈缺电子状态,而CoP中P带部分负电荷呈电子富集状态,均匀分布于催化剂中的B也呈缺电子状态.换而言之,有部分电子从B和Co转移到P.
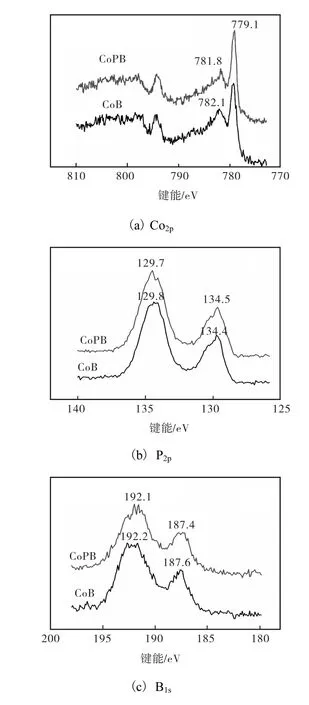
图4 使用过的催化剂XPS图谱Fig.4 XPS spectra of spent catalysts in different energy levels
2.5 催化剂活性测试及分析
2.5.1 反应温度对催化剂活性的影响及活性相的确定
图5显示了不同温度下CoB/CNTs和CoBP/ CNTs(n=0.43)的 PH3分解活性,其中 PH3空速为2,520,mL/(h·gcat).2个样品的催化活性都随温度升高而逐渐增强,这是由于 PH3分解为吸热反应[13],反应温度升高有利于反应的进行.当温度达到 400,℃时,CoBP/CNTs(n=0.43)和 CoB/CNTs的分解率分别达到了99.8%和 89.1%.当温度升高到 440,℃时,CoBP/CNTs(n=0.43)的分解率基本保持不变,为99.9%,而CoB/CNTs的分解率升高到99.8%.活性测试结果表明,CoBP/CNTs(n=0.43)的催化活性高于CoB/CNTs.前述 XRD 检测表明,使用过的 CoBP/CNTs(n=0.43)样品中,只有CoP相出现;而反应后的CoB/CNTs样品中,除了CoP相以外,还有金属Co.将XRD与催化剂活性检测结果相结合,可以推测出:对于PH3分解反应,钴的磷化物活性高于金属钴.金属磷化物是P填隙在金属原子晶格内而形成的化合物.当非金属元素填隙于过渡金属晶体内时,过渡金属的结构和外层电子结构在微观上会发生改变,导致其对反应物的吸附和活化能力改善.Sun等[14]认为PH3分解的机理分为以下3个步骤:首先,PH3与催化剂表面发生可逆的化学吸附;然后,活化的 PH3发生P—H键断裂,连续解离脱氢;最后P原子和H原子分别结合成P2和H2脱附.现有对PH3分解的研究认为,P—H键断裂为反应的限速步骤[14-15].前述 XPS检测表明,样品催化过程形成的CoP相中,Co电子密度较正常状态小,非晶态合金晶化后,金属的电子密度为正常状态[12].对于反应中出现的CoP和金属Co而言,CoP中 Co的电子密度要小于金属 Co的电子密度,当PH3与 Co形成配位键时,前者的Co—P键强度要大于后者,更有利于 P—H键的断裂.因此,反应过程中,全部为CoP相的CoBP/CNTs(n=0.43)活性高于有部分Co存在的CoB/CNTs.

图 5 制备的 CoB/CNTs和 CoBP/CNTs(n=0.43)催化剂在不同温度下对PH3的分解效果Fig.5 PH3 decomposition on as-prepared CoB/CNTs and CoBP/CNTs(n=0.43)catalysts at different temperatures
2.5.2 P与(B+P)摩尔比对催化剂活性的影响
图 6为 CoB/CNTs和不同 n的 CoBP/CNTs在300~440,℃温度范围内反应8 h后,降温至 360,℃,对PH3分解的活性,PH3空速为2,520,mL/(h·geat).由图6可见,在CoB/CNTs中添加P可加强PH3分解活性.如前所述,P的加入可使催化剂中 Co更好地转化为活性更高的 CoP相.当 n=0.43时,CoBP/CNTs的活性达到最高.但随着 P加入量的增加,CoBP/CNTs的催化活性提高程度又有所降低,这可能与制备样品颗粒的大小有关.如图 1所示,相同方法制备的CoPB/CNTs(n=0.43)颗粒要大于CoB/CNTs,这是因为 BH4-的还原能力远强于 H2PO2-,当仅用 H2PO2-还原 Co盐时,所得的 CoP非晶态颗粒要比CoB大得多[7].PH3分解反应发生于这类较大颗粒的非晶态合金上时,形成的 CoP化合物颗粒也会大于较小颗粒上形成的CoP化合物颗粒.因此,对于采用BH4-和(或)H2PO2-还原 Co离子制备的催化PH3分解的材料来说,仅用 BH4-作为还原剂制备的CoB颗粒小,但催化反应过程中,金属原子不易充分磷化成具有更高活性的 CoP化合物,因而催化活性不高;而用过多 H2PO2-取代 BH4-作为还原剂制备的CoBP非晶态合金,虽然在反应过程中金属 Co较易于磷化,但由于反应中形成的CoP化合物较大,减少了活性相 CoP与反应物接触的面积,降低了催化相活性位的有效利用率,因而催化活性仅较由 CoB产生的催化剂有轻微提高.当 CoBP/CNTs中 n=0.43左右时,催化剂颗粒大小与活性相CoP化合物形成2个因素的综合作用达到最佳状态,PH3分解率最高.

图 6 制备的 CoB/CNTs和 CoBP/CNTs催化剂中 P含量与PH3分解效果的关系Fig.6 Relationship of P content and PH3 decomposition on as-prepared CoB/CNTs and CoBP/CNTs catalysts
3 结 语
用诱导-化学还原法制备了一系列 CNTs负载的CoB和不同P与(B+P)摩尔比的CoBP非晶态合金,并用于 PH3催化分解反应.结果显示:在 CoB/CNTs制备过程中加入次磷酸盐制备的CoBP/CNTs催化活性提高,最高催化活性为 CoBP中 n=0.43左右,增加CoBP中P含量可使得 CoBP/ CNTs在催化过程中生成活性更高的 CoP金属填隙化合物,而非晶态CoB/CNTs在催化过程中,除形成 CoP相外,还存在部分活性较低的金属 Co.当 CoBP中 n大于 0.58时,因为 H2PO2-较弱的还原能力,使得制备的非晶态颗粒增大,进而导致反应过程中生成的 CoP活性相颗粒增大,P添加对CoB/CNTs催化活性的促进作用又有所降低.
[1] Zhang Rui,Wu Min,Wang Qing,et al. The determination of atmospheric phosphine in Ny-Ålesund[J]. Chinese Science Bulletin,2010,55(16):1662-1666.
[2] 余琼粉,易红宏,唐晓龙,等. 磷化氢净化技术及其展望[J]. 环境科学与技术,2009,32(10):87-91.Yu Qiongfen,Yi Honghong,Tang Xiaolong,et al.Progress on phosphine control technology[J]. Environmental Science and Technology,2009,32(10):87-91(in Chinese).
[3] ISO A. Preparation of high-purity phosphorus:JP,602215510[ P]. 1985-10-28.
[4] Li Lili,Han Changxiu,Yang Li,et al. The nature of PH3decomposition reaction over amorphous CoNiBP alloy supported on carbon nanotubes[J]. Industrial and Engineering Chemistry Research,2010,49(4):1658-1662.
[5] Han Changxiu,Liu Shuangxi,Ren Jili,et al. Decomposition of PH3to high purity phosphorus over Co-B amorphous alloy[J]. Journal of Molecular Catalysis,2009,23(1):1-5.
[6] Wonterghem J V,Morup S M,Koch C J,et al. Formation of ultra-fine amorphous alloys particles by reduction in aqueous solution[J]. Nature,1986,322:622-623.
[7] Li Hui,Yang Pingfeng,Chu Dongsheng,et al. Selective maltose hydrogenation to maltitol on a ternary Co-PB amorphous catalyst and the synergistic effects of alloying B and P[J]. Appllied Catalysis A:General,2007,325:34-40.
[8] Cecilia J A,Infantes-Molina A,Rodríguez-Castellón E,et al. Dibenzothiophene hydrodesulfurization over cobalt phosphide catalysts prepared through a new synthetic approach:Effect of the support[J]. Applied Catalysis B:Environmental,2009,92:100-113.
[9] Izhar S,Nagai M. Transition metal phosphide catalysts for hydrogen oxidation reaction[J]. Catalysis Today,2009,146(1/2):172-176.
[10] Sawhill S J,Layman K A,Van Wyk D R,et al. Thiophene hydrodesulfurization over nickel phosphide catalysts:Effect of the precursor composition and support[J]. Journal of Catalysis,2005,231:300-313.
[11] Wang R,Smith K J. Hydrodesulfurization of 4,6-dimethyldibenzothiophene over high surface area metal phosphides[J]. Applied Catalysis A:General,2009,361:18-25.
[12] Li Hui,Li Hexing,Deng Jingfa. The crystallization process of ultrafine Ni-B amorphous alloy[J]. Materials Letters,2001,50:41-46.
[13] Jacobson M L,Chiu M C,Crowell J E. P2desorption from phosphine decomposition on Si(100)surfaces[J].Langmuir,1998,14(6):1428-1434.
[14] Sun Y,Law D C,Hicks R F. Kinetics of phosphine adsorption and phosphorus desorption from gallium and indium phosphide(001)[J]. Surface Science,2003,540(1):12-22.
[15] Fu Q,Li L,Li C H,et al. Mechanism of arsine adsorption on the Gallium-rich GaAs(001)-(4×2)surface[J]. Journal of Physical Chemistry B,2000,104(23):5595-5602.
猜你喜欢
电镀与精饰(2022年10期)2022-10-14
电镀与精饰(2022年3期)2022-03-14
云南化工(2020年11期)2021-01-14
矿产综合利用(2020年1期)2020-07-24
机械制造(2020年5期)2020-02-20
表面工程与再制造(2019年6期)2019-08-24
湖南大学学报·自然科学版(2017年12期)2018-01-17
物理学报(2017年17期)2017-09-09
中国科技纵横(2016年19期)2016-12-10
新疆钢铁(2015年3期)2015-11-08
