
异氟烷心肌保护的分子机制的初步研究
2011-06-22 01:03刘志刚刘永芳陈雪君赵博吴迪周芳
东南大学学报(医学版) 2011年5期
刘志刚,刘永芳,陈雪君,赵博,吴迪,周芳
(1.武汉大学人民医院麻醉科,湖北 武汉 430060;2.武汉市第一医院 麻醉科,湖北武汉 430060)
心肌缺血再灌注损伤(myocardial ischemia reperfusion injury,MIRI)是临床上各种疾病、休克、创伤及体外循环心血管手术等过程中常见的损伤,由多种细胞内信号途径参与并介导。研究发现,血管内皮生长因子(vascular endothelial growth factor,VEGF)与心肌缺血再灌注损伤具有密切关系。VEGF是一种分泌性糖蛋白,可经缺血缺氧诱导产生,与其受体结合后具有促进内皮细胞增生、迁移,增加血管通透性和加速新血管形成的作用,并对缺血心肌具有保护效应[1]。
Kawata等[2]的研究表明,吸入麻醉药异氟醚预处理的心肌保护作用与VEGF表达上调有关,其中蛋白激酶C(protein kinase C,PKC)家族是介导预适应保护作用的重要途径,但其具体的作用机制不明。本实验通过观察临床常用吸入性麻醉药异氟烷对培养大鼠心肌细胞VEGF分泌及PKC亚型表达的影响,旨在进一步探讨异氟烷心肌保护的分子机制,为围手术期心肌保护提供理论依据。
1 材料与方法
1.1 主要试剂与药品
PKC抑制剂calphostin C(Sigma公司,美国),5'-溴脱氧尿苷(Sigma公司,美国),异氟烷(Abbott公司,美国),高糖DMEM培养基(Hyclone公司,美国),胎牛血清(Hyclone公司,美国),兔抗鼠 PKCα、PKCδ、PKCζ和PKCε抗体(Santa Cruz公司,美国),羊抗兔 IgG(Santa Cruz公司,美国),PVDF蛋白印迹膜(Bio-Rad公司,美国),化学发光试剂盒(Pierce公司,美国),BCA蛋白质定量试剂盒(Pierce公司,美国),VEGF酶联免疫试剂盒(R&D Systems公司,美国),其他试剂均为Sigma公司产品。
1.2 心肌细胞分离培养
取1~3 d新生SD大鼠,雌雄不拘,对文献[3]的方法略作改良来分离培养心肌细胞。无菌条件下开胸取出心室肌,清洗后加入0.05%胶原酶、0.08%胰蛋白酶和0.02%EDTA的消化液,分次消化、离心,以15%胎牛血清的高糖DMEM悬浮细胞,差速贴壁法去除成纤维细胞。将收集心肌细胞的浓度调整为5×105ml-1之后均匀接种,置CO2培养箱(体积分数为5%的CO2,37℃)中培养。培养的前2 d加入5-溴脱氧嘧啶 0.1 mmol·L-1,抑制非心肌细胞贴壁生长。
1.3 实验分组
心肌细胞随机分为:对照组(CON组),细胞不经任何处理;异氟烷组(ISO组),将细胞培养皿置入设有通气及出气接口的自制小无菌密闭容器中,保持37℃恒温,容器进气端与麻醉机相连,持续输入经气体监护仪校正的异氟烷,以维持容器内异氟烷体积分数为1.4%,6 h后关闭异氟烷;PKC抑制剂组(CAL组),PKC抑制剂calphostin C加入细胞培养液中使其终浓度为50 nmol·L-1;PKC抑制剂+异氟烷组(IC组),心肌细胞培养液中加入 calphostin C 50 nmol·L-1,经1.4% 的异氟烷共同作用 6 h[4]。
1.4 指标测定
(1)培养液VEGF:收集细胞培养液经1 500 r·min-1离心5 min,取上清成分,采用ELISA方法检测VEGF的含量。根据试剂盒要求依次加样、孵育、洗涤、显色,最后在酶标仪450 nm波长测定样品的吸光度。每份样本设2个复孔。以吸光度值为纵坐标,标准品质量浓度(pg·ml-1)为横坐标,绘制标准曲线,求出标准方程。通过标本的吸光度值得出培养上清液中的VEGF浓度。(2)心肌细胞PKC蛋白表达:参照Kanaya等[5]介绍的方法进行,分别提取心肌细胞细胞质成分和细胞膜成分的蛋白,用BCA方法进行蛋白定量。用SDS-PAGE电泳分离并电转移至PVDF膜上。将PVDF膜以含3%牛血清蛋白的TBS室温封闭1 h,然后分别加入1∶500 PKCα、PKCδ、PKCζ和 PKCε一抗4℃孵育过夜,漂洗后加入1∶1 000的二抗室温作用1 h。经ECL试剂盒进行发光显影于X线胶片上,采用凝胶成像系统分析处理。蛋白表达量以条带的吸光度值×面积计算,以GAPDH作为内参对照校正上样量,结果以各组PKC含量相对CON组PKC膜蛋白表达比值表示。
1.5 统计学处理
2 结 果
2.1 PKC抑制剂抑制异氟烷诱导的VEGF分泌
经1.4%异氟烷作用6 h后,CON、ISO、CAL和IC组的 VEGF含量分别为(62.67±6.713)、(101±13.27)、(64.67 ± 10.78)和(76.00 ± 7.43)pg·ml-1。与CON组比,ISO组VEGF含量显著升高(P<0.01);经PKC抑制剂calphostin C处理后,VEGF含量显著降低,明显低于ISO组(P<0.01);CAL组与CON组比较差异无统计学意义(P>0.05)。
2.2 异氟烷诱导PKCε的激活转位
经1.4%的异氟烷作用6 h后,与CON组比,ISO组PKCα、PKCδ和PKCζ亚型在细胞质和细胞膜表达均无统计学意义(P>0.05),细胞质中3种PKC含量均高于细胞膜中,但ISO组PKCε亚型细胞膜浓度显著升高,而细胞质浓度显著降低(P<0.01)(表1、图1)。
表1 细胞膜和胞质中4种PKC含量的比较±s)Tab 1PKC isoforms expressions in the membrane and the cytosol±s)
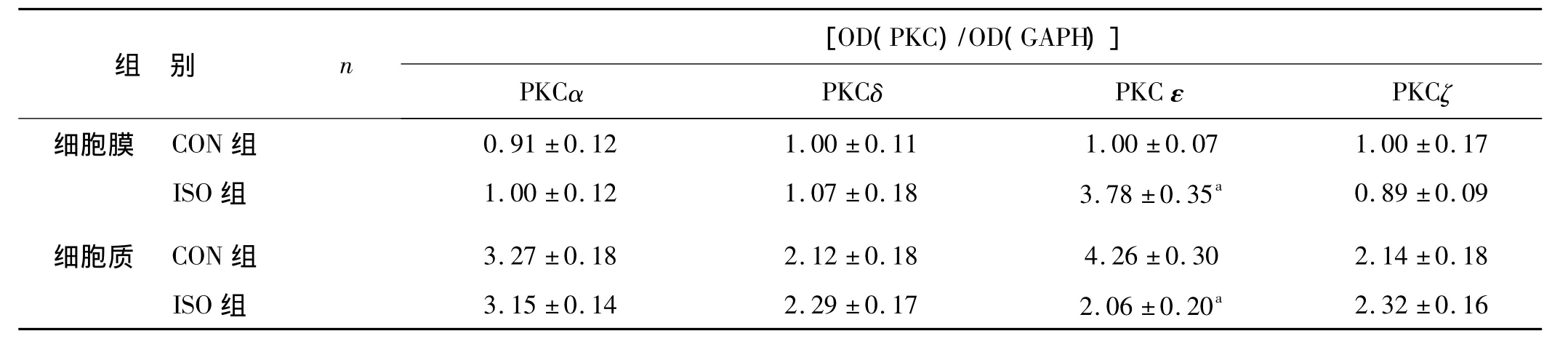
表1 细胞膜和胞质中4种PKC含量的比较±s)Tab 1PKC isoforms expressions in the membrane and the cytosol±s)
与相应CON组比,a P<0.01
组 别 n [OD(PKC)/OD(GAPH)]PKCα PKCδ PKC ε PKCζ细胞膜 CON组0.91 ±0.12 1.00 ±0.11 1.00 ±0.07 1.00 ±0.17 ISO 组 1.00 ±0.12 1.07 ±0.18 3.78 ±0.35a 0.89 ±0.09细胞质 CON 组 3.27 ±0.18 2.12 ±0.18 4.26 ±0.30 2.14 ±0.18 ISO 组 3.15 ±0.14 2.29 ±0.17 2.06 ±0.20a 2.32 ±0.16
3 讨 论
心肌缺血再灌注损伤是临床上各种疾病和治疗过程中常见的病理损伤。心肌血管生成是心肌保护的重要机制之一,梗死区内是否有血管迅速生成,与心肌梗死后能否建立良好的侧支循环以改善梗死区血液供应及梗死区心肌的存活均密切相关[6]。因此,及时采取有效干预措施来促进缺血区的新血管生成,对心肌缺血再灌注损伤后心功能的恢复具有非常重要意义。
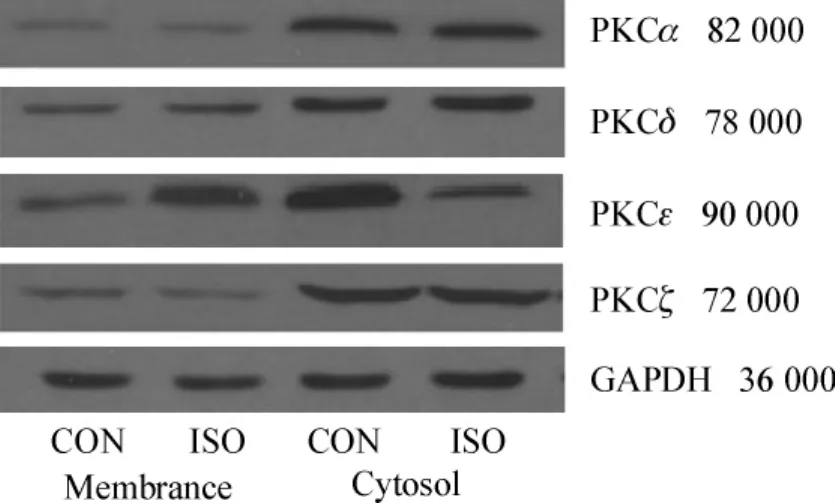
图1 4种PKC亚型细胞质和细胞膜PKC含量的Western blotting结果Fig 1 PKC isoforms expressions detected by Western blotting in the membrane and the cytosol
VEGF是迄今发现的特异性最高、活性最强的血管生长因子,可促进内皮祖细胞的增殖、迁移和趋化。在正常情况下,心肌细胞内的VEGF含量很少,用一般的检测方法很难发现。当冠状动脉狭窄或梗塞导致心肌缺血缺氧时,可刺激局部心肌迅速表达VEGF基因及其蛋白。VEGF表达受细胞内蛋白激酶系统(如PKC)等多个不同环节的调控[7]。Kawata 等[2]发现,吸入麻醉药异氟醚预处理心肌的保护作用与VEGF表达上调有关,其中蛋白激酶C家族通路是介导预适应保护作用的重要途径。本研究显示,当心肌细胞经1.4%的异氟醚作用6 h后,与 CON组比较,ISO组VEGF值显著升高,CAL组和IC组VEGF值差异无统计学意义,说明PKC抑制剂calphostin C预处理阻断了isoflurane诱导的VEGF释放,但calphostin C对心肌细胞VEGF分泌无显著影响。
PKC是一种由钙离子激活的磷脂依赖性蛋白激酶,是细胞内信号传导的重要递质。PKC存在于多种组织中,迄今发现至少12种以上的PKC亚型。PKC分为细胞质和细胞膜两部分,生理条件下细胞质PKC没有活性,当受到一定条件的刺激后,细胞质PKC转位到细胞膜。因而,细胞膜及细胞质PKC含量的动态变化可作为酶活化的标志。在心肌细胞,仅有PKCα、PKCδ、PKCζ和 PKCε 4种亚型表达。近年 Pravdic等[8-9]报道PKC活化后降低缺血再灌注心肌细胞凋亡,认为PKC活化后其亚型PKCε可由细胞质向细胞膜的转位,最终导致细胞膜ATP依赖的钾通道(KATP)的开放,从而减少心肌缺血再灌注损伤。本实验通过对4种PKC亚型的研究发现,ISO组PKCα、PKCδ和PKCζ亚型在细胞质和细胞膜差异均无统计学意义,细胞质浓度高于细胞膜,但ISO组PKCε亚型细胞膜浓度显著升高,而细胞质浓度显著降低,说明Isoflurane诱导心肌细胞PKCε从细胞质向细胞膜转位,但对PKC其它亚型的表达没有影响。
目前关于麻醉药心血管保护作用的研究,主要限于麻醉药对心肌缺血再灌注损伤的近期保护效应(再灌注0~72 h),而对心肌缺血再灌注损伤的远期效应及其分子机制尚未见报道。本研究发现,临床相关浓度异氟烷通过PKCε转位上调心肌细胞VEGF表达,将为吸入麻醉药的远期器官保护效应提供新的思路。VEGF上调可诱导内皮祖细胞增殖、迁移和趋化,促进缺血损伤心肌血管新生,改善缺血再灌注损伤后心室重构[10]。
综上所述,异氟烷通过蛋白激酶PKCε途径诱导心肌细胞分泌VEGF,可能是异氟烷心肌保护的分子机制之一。
[1]DARGIE H.Heart failure post-myocardial infarction:a review of the issues[J].Heart,2005,91(Suppl 2):ii3-6.
[2]KAWATA H,YOSHIDA K,KAWAMOTO A,et al.Ischemic preconditioning upregulates vascular endothelial growth factor mRNA expression and neovascularization via nuclear translocation of protein kinase C epsilon in the rat ischemic myocardium[J].Circ Res,2001,88(7):696-704.
[3]JAMNICKI-ABEGG M,WEIHRAUCH D,PAGEL P S,et al.Isoflurane inhibits cardiac myocyte apoptosis during oxidative and inflammatory stress by activating Akt and enhancing Bcl-2 expression[J].Anesthsiology,2005,103(5):1006-1014.
[4]OBAL D,WEBER N C,ZACHAROWSKI K,et al.Role of protein kinase C-epsilon(PKC epsilon)in isoflurane-induced cardioprotection[J].Br J Anaesth,2005,94(2):166-173.
[5]KANAYA N,GABLE B,MURRAY P A,et al.Propofol increases phosphorylation of troponin I and myosin light chain 2 via protein kinase C activation in cardiomyocytes[J].Anesthesiology,2003,98(6):1363-1371.
[6]NEUFELD G,COHEN T,GENGRINOVITCH S,et al.Vascular endothelial growth factor(VEGF)and its receptors[J].FASEB J,1999,13(1):9-22.
[7]UECKER M,da SILVA R,GRAMPP T,et al.Translocation of protein kinase C isoforms to subcellular targets in ischemic and anesthetic preconditioning[J].Anesthesiology,2003,99(1):138-147.
[8]PRAVDIC D,SEDLIC F,MIO Y,et al.Anesthetic-induced preconditioning delays opening of mitochondrial permeability transition pore via protein kinase C-epsilon-mediated pathway[J].Anesthesiology,2009,111(2):267-274.
[9]LUDWIG L M,WEIHRAUCH D,KERSTEN J R,et al.Protein kinase C translocation and Src protein tyrosine kinase activation mediate isoflurane-induced preconditioningin vivo:potential downstream targets of mitochondrial adenosine triphosphate-sensitive potassium channels and reactive oxygen species[J].Anesthesiology,2004,100(3):532-539.
[10]GRUNEWALD M,AVRAHAM I,DOR Y,et al.VEGF-induced adult neovascularization:recruitment,retention,and role of accessory cells[J].Cell,2006,124(1):175-189.
东南大学学报(医学版)2011年5期