
HPLC同时测定米格列醇片有关物质和含量的方法改进研究*
2011-06-21 02:16郝光启郁树涛孙彦余
天津药学 2011年2期
郝光启,郁树涛,孙彦余
(鲁南制药集团股份有限公司,临沂 276006)
米格列醇(C8H17NO5)为去氧野尻霉素衍生物,是一种新型口服α-葡萄糖苷酶抑制剂。如按国家食品药品监督管理局颁布的质量标准(YBH00592010)对该药进行检测,会出现的诸多问题而无法正常检测[1]。本文通过研究,改变了原流动相组成及pH值,建立了新方法,解决了上述问题。该方法专属性强、灵敏度高、准确度和重复性好、方法简便,可同时用于米格列醇片有关物质和含量的定量测定。
1 仪器与试药
1.1仪器 Agilent 1100高效液相色谱系统:G1379A脱气机、G1311A四元泵、G1313A自动进样器、G1316A柱温箱、G1315B DAD检测器、A 10 01化学工作站(美国安捷伦公司),SHH-100GD药品强光照射试验箱(重庆市永生实验仪器厂),YB系列真空恒温干燥箱(天津药典标准仪器厂),TTL-10B超纯水系统(北京同泰联科技发展有限公司),PHS-3C型精密pH计(上海精密科学仪器有限公司),XS204型电子天平(瑞士梅特勒-托利多公司)。
1.2试药 米格列醇对照品(本公司生产的原料药,经HPLC面积归一化法测定纯度为99.8%以上),米格列醇片(本公司生产,批号:081201、081202、081203)。乙腈为色谱纯(山东禹王实业有限公司化工分公司),磷酸二氢铵、三乙胺均为分析纯,实验用水为超纯化水。
2 方法与结果
2.1色谱条件及系统适用性 色谱柱:Lichrospher氨丙基硅烷键合硅胶色谱柱(250 mm×4.6 mm,5 μm)(江苏汉邦科技有限公司);流动相:乙腈-0.01 mol/L磷酸二氢铵溶液(三乙胺调pH至7.5)(65∶35);检测波长:210 nm;流速:1.0 ml/min;进样量:20 μl;柱温:25 ℃;检测器:二极管阵列检测器(DAD)。理论塔板数按米格列醇峰计算应不低于1 000。
2.2溶液制备
2.2.1有关物质供试品溶液 取本品细粉适量(含米格列醇约200 mg),精密称定,加流动相溶解并稀释至100 ml,精密量取此液,用流动相稀释成每1 ml含有600 μg的溶液,摇匀,滤过,取续滤液作为供试品溶液。
2.2.2有关物质对照溶液 精密量取供试品溶液1 ml,置100 ml量瓶中,加流动相稀释至刻度,摇匀,即得。
2.2.3含量测定供试品溶液 取本品20片,精密称定,研细,混合均匀;精密称取适量(相当于米格列醇约100 mg),加流动相适量溶解并稀释成每1 ml中含米格列醇约100 μg,摇匀,滤过。
2.2.4含量测定对照品溶液 取经60 ℃减压干燥至恒重的米格列醇对照品适量,精密称定为10.6 mg,置50 ml量瓶中,加流动相溶解并稀释至刻度,摇匀,作为对照品储备液。精密量取溶液5 ml置于10 ml量瓶中,加流动相稀释至刻度,摇匀,即得对照品溶液。
2.3专属性试验
2.3.1空白辅料干扰试验 取米格列醇片空白辅料适量(约相当于200 mg的米格列醇处方量),按有关物质供试品制备法配制溶液,照上述色谱条件依法测定,结果见图1。由图可见,空白辅料对米格列醇的测定无干扰。
2.3.2样品破坏性试验
2.3.2.1酸破坏 取样品适量(相当于米格列醇约15 mg),置25 ml量瓶中,加0.1 mol/L盐酸溶液5 ml,100 ℃水浴加热5 h,冷却,加碱中和后,加流动相稀释至刻度,摇匀,滤过,取续滤液依法检查。
2.3.2.2碱破坏 取样品适量(相当于米格列醇约15 mg),置25 ml量瓶中,加0.1 mol/L氢氧化钠溶液5 ml,100 水浴加热3 h,冷却,加酸中和后,加流动相稀释至刻度,摇匀,滤过,取续滤液依法检查。
2.3.2.3热破坏 取样品适量(相当于米格列醇约15 mg),置25 ml量瓶中,于140 ℃恒温箱中放置5d,冷却,加流动相溶解并稀释至刻度,摇匀,滤过,取续滤液依法检查。
2.3.2.4氧化破坏 取样品适量(相当于米格列醇约15 mg),置25 ml量瓶中,加30%过氧化氢溶液0.5 ml,常温放置1 min,加流动相稀释至刻度,摇匀,滤过,取续滤液依法检查。
2.3.2.5光照破坏 取样品适量(相当于米格列醇约15 mg),置25 ml量瓶中,加流动相溶解并稀释至刻度,摇匀,置于照度为4 500 Lx强光照射试验箱中照射20 d,滤过,取续滤液依法检查。
强制降解结果表明:本方法专属性强,米格列醇与破坏后的辅料峰及降解产物完全分离,说明本色谱条件适用于米格列醇片的检测。色谱图见图1。
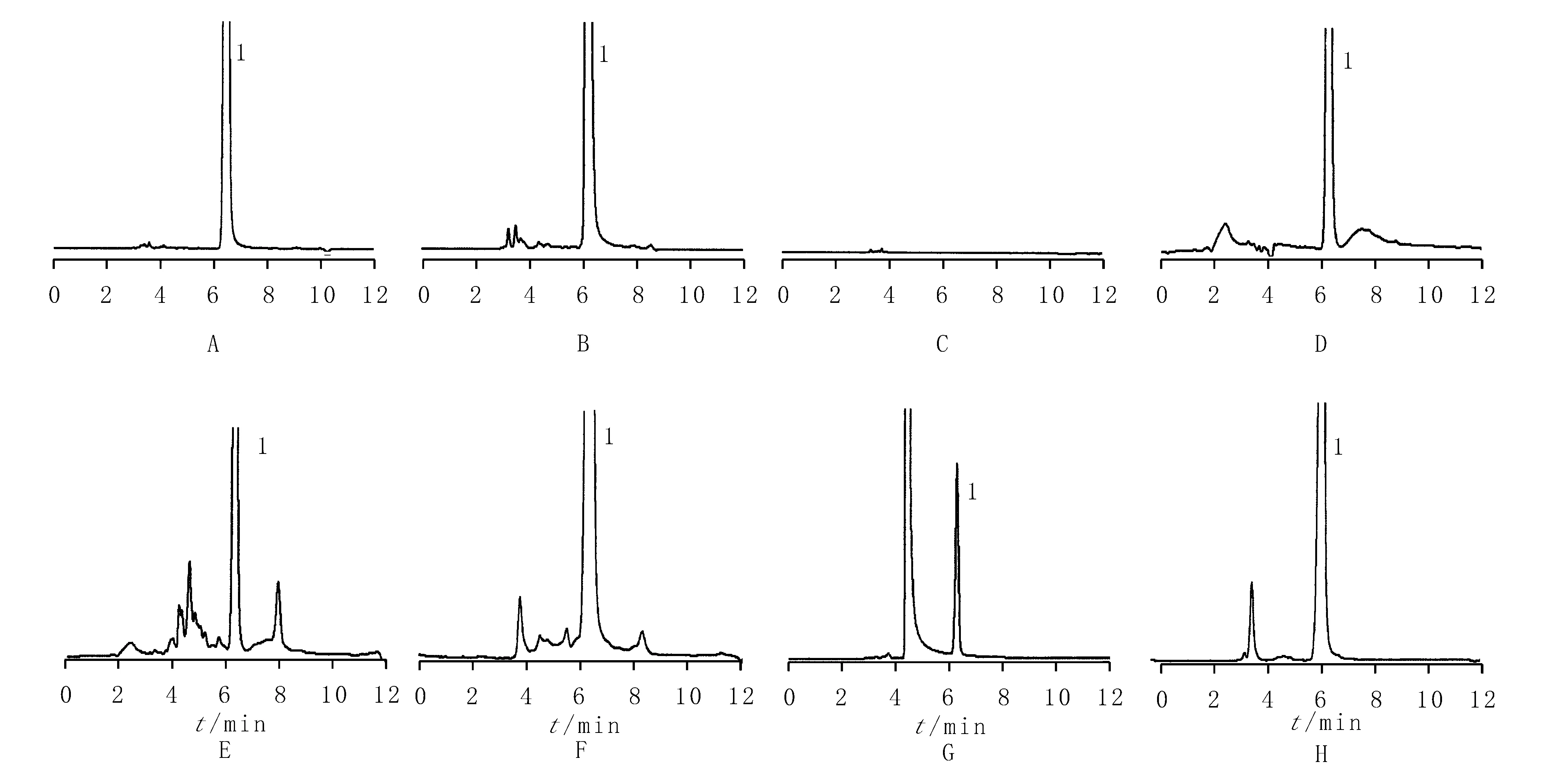
1.米格列醇
2.4有关物质检查 依“2.2.1”和“2.2.2”项下方法分别配制有关物质供试品溶液和对照溶液。精密量取对照溶液20 μl注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的10%~20%;再准确量取供试品溶液20 μl注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍。供试品溶液色谱图中如显示杂质峰,扣除辅料峰外,单个杂质峰面积不得大于对照溶液主峰面积的1/2(0.5%),各杂质峰峰面积的和,不得大于对照溶液主峰面积(1.0%)。三批样品(批号081201、081202、081203)最大单个杂质结果分别为:0.08%、0.08%和0.07%,总有关物质分别为:0.60%、0.55%和0.51%,结果均符合规定。
2.5含量测定
2.5.1线性试验 取“2.2.4”项下对照品储备液,分别精密量取1.0、3.0、5.0、7.0和9.0 ml,各置于10 ml量瓶中,加流动相稀释至刻度,摇匀。依法测定,以米格列醇峰面积对其浓度(μg/ml)进行线性回归,回归方程为:Y=0.13X-3.13(r=0.999 5)。由此可见:米格列醇浓度在21.2~190.8 μg/ml范围内线性关系良好。
2.5.2精密度试验 取“2.2.4”项下对照品溶液,依法连续进样6次,记录峰面积,结果米格列醇峰面积的RSD为0.074%(n=6),说明方法重复性良好。
2.5.3重复性试验 取一批样品(批号:081201),分别依“2.2.3”和“2.2.4”项下方法配制6份供试品溶液和1份对照品溶液,在确定的色谱条件下测定,米格列醇片的平均含量(占标示量)为99.1%,RSD为0.083%(n=6)。
2.5.4溶液稳定性试验 取“2.2.3”项下含量测定供试品溶液,依法分别于0、1、3、5、8和10 h后进样分析。结果米格列醇峰面积的RSD为0.13%(n=6),表明米格列醇溶液在10 h内稳定。
2.5.5回收率试验 分别精密称取经60 ℃减压干燥至恒重的米格列醇对照品16、20和24 mg各3份,各置于100 ml量瓶中,并按处方量比例分别加入空白辅料,加流动相溶解并稀释至刻度,然后依“2.2.4”项下对照品溶液配制方法制备9份回收率试验溶液。取“2.2.4”项下对照品溶液,按外标法以峰面积计算依法测定,结果平均回收率为100.1%,RSD为1.07%,见表1。

表1 含量回收率试验结果(%,n=9)
2.5.6定量限和检测限试验 精密称取米格列醇对照品适量,用流动相溶解并稀释制成一系列不同浓度的溶液,依法测定。当信噪比S/N =10时,米格列醇浓度为0.19 μg/ml,米格列醇的定量限为3.80 ng;当信噪比S/N =3时,米格列醇浓度为0.062 μg/ml,米格列醇的检测限为1.24 ng。
2.5.7含量测定结果 取3批样品(批号081201、081202、081203)依法测定,结果三批样品含量(占标示量)分别为99.1%、98.6%和100.3%,均符合规定。
3 讨论
依国家药监局批准的米格列醇片质量标准(YBH00592010),如前言所述情况,该产品无法正常检测,而本文建立的方法专属性强、准确度高、重复性好,可同时用于米格列醇片的有关物质检查和含量测定,经实验证实本法亦可作为米格列醇原料药的有关物质检查和含量测定的检测方法。
1 郝光启,李进启,葛梅,等.米格列醇片溶出度HPLC检测方法改进研究.天津药学,2011,23(1): 16
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
临床医药文献杂志(电子版)(2020年75期)2021-01-21
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
小哥白尼(军事科学)(2018年2期)2018-05-25
数学小灵通(1-2年级)(2017年9期)2017-10-13
百科探秘·航空航天(2017年6期)2017-07-10
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
百科探秘·航空航天(2016年10期)2016-02-27
天津药学(2013年2期)2013-12-23
