
吲达帕胺片体外溶出度考查*
2011-06-21 02:41唐素芳高立勤杭太俊
天津药学 2011年3期
王 昕,唐素芳,高立勤,杭太俊
(1.天津市药品检验所,天津 300070; 2.中国药科大学,南京 210009)
吲达帕胺(Indapamide)是一种具有降压利尿双重作用的非噻嗪类吲哚衍生物,通过抑制远端肾小管皮质稀释段的再吸收水与电解质而发挥作用。该药以其降压作用持久、毒副作用小、价格低廉等特点受到广大高血压患者的青睐,是临床用于抗高血压治疗的常用药之一。目前,国内有多个厂家生产吲达帕胺片,各厂家生产工艺的技术含量不同,其产品的内在质量也不同。
吲达帕胺片在《中国药典》和进口药品注册标准中均有收载。其中,《中国药典》2005年版二部[1]采用乙醇-水(5∶895)作为溶出介质。不难看出,由于本品的难溶解性使得在溶出介质中使用了有机溶剂以期达到满意的溶出曲线,但这与人体的实际生理情况相差很大;《中国药典》2010年版二部[2]将溶出介质修订为pH 6.8的磷酸盐缓冲液;进口药品注册标准(J×19990200)采用0.1 mol/L盐酸溶液作为溶出介质。对于一个理想的药物来说,无论患者体内的胃酸水平是否正常、亦无论患者是身强力壮的年轻人还是胃肠蠕动功能较弱的老年人都应该具有一定的、适当的体外溶出行为,才能保证患者服药后有一定的疗效和作用。为此,本试验选择上述现行两方法及参照日本“药品品质再评价”工程中推荐使用的模拟人体消化道环境的4种溶剂和条件[3]测定溶出度,旨在考查国产与进口吲达帕胺片的内在质量。
1 仪器与药品
1.1仪器 AT-TSmart 药物溶出度仪(美国SOTAX公司);UV-2450紫外分光光度计(日本岛津公司);LC-2010C高效液相色谱仪(Agilent公司)。
1.2试药 吲达帕胺对照品(中国药品生物制品检定所提供,纯度99.5%);吲达帕胺片A厂(法国施维雅药厂,批号835867);B厂(新疆,批号20091023);C厂(河北省,批号090602);D厂(湖北省,批号20091001)。甲醇为色谱纯,甲酸、冰醋酸、盐酸、磷酸二氢钾、磷酸氢二钠、醋酸钠、氯化钠和氢氧化钠均为分析纯。
2 试验条件的选择
2.1溶出度试验条件的确定 考虑患者年龄、体质、身体状况等的不同,本试验采用现行的进口药品注册标准、《中国药典》2010年版方法及日本“药品品质再评价工程”中推荐使用的四种溶剂(以下分别简称为方法1~6)分别测定。方法为取本品,照溶出度测定法(《中国药典》2010年版二部附录Ⅹ C),方法、转速、溶出介质见表1,依法操作,取样时间为5、10、15、30、45、60和90 min及2、3、4、5和6 h(2 h后连续两个取样时间点的溶出量达到90%时可提前结束),取溶液3 ml(同时补液),滤过,取续滤液作为供试品溶液;另取吲达帕胺对照品25 mg,精密称定,置50 ml量瓶中,加甲醇溶解并定容至刻度,摇匀,精密量取适量,用溶出介质稀释制成每1 ml中含吲达帕胺约为5 μg或2.5 μg的溶液,作为对照品溶液,测定方法见表1。
2.2溶出度测定方法的建立 方法1和2分别采用紫外分光光度法中的双波长(A240-A275)和单波长(A240)法测定,日本再评价方法只提供了模拟人体消化液的4种溶出介质。经试验,采用各生产厂提供的处方配制辅料溶液(采用各方法项下的溶剂制备),经紫外吸收图谱的扫描可以看出,辅料溶液在本品的最大吸收波长240 nm处有紫外吸收,由于供试液自身的吸收度值偏低,故辅料对测定有一定的影响,且除方法2中A240和A275近似为等吸收外,其他方法两吸收值均有一定差距。为使测定结果准确,建立专属性更强的HPLC法进行测定。

表1 溶出度试验条件
2.2.1色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,甲醇-水-甲酸(55∶45∶0.1)为流动相,检测波长为240 nm,进样20 μl,流速1.0 ml/min,理论板数按吲达帕胺峰计算为5 063。在该色谱条件下,辅料溶液对测定无影响,被测成分峰保留时间适宜,与相邻杂质峰可达基线分离,结果满意。色谱图见图1。图1中A图1-6分别代表表1 方法1~6测得结果。

1.吲达帕胺
2.2.2线性关系考查 取吲达帕胺对照品约12.5 mg,精密称定,置250 ml量瓶中,加甲醇溶解并定容至刻度,摇匀,精密量取该溶液1、2、4、6、8、10和20 ml,分别置200 ml量瓶中,用各自的溶出介质定容至刻度,摇匀,精密量取上述溶液各20 μl,分别注入液相色谱仪,记录色谱图,以浓度C为横坐标,以峰面积A为纵坐标,绘制A-C曲线,结果表明,吲达帕胺在0.250 6~5.012 μg/ml浓度范围内线性关系良好,见表2。

表2 线性试验测定结果
2.2.3回收率试验 取吲达帕胺对照品约25 mg,精密称定,置50 ml量瓶中,加甲醇溶解并定容至刻度,摇匀,作为对照品储备液。精密量取对照品储备液3、4和5 ml各3份置50 ml量瓶中,分别加入1片的辅料量,用各自的溶出介质稀释至刻度,摇匀,再分别精密量取5 ml置50 ml量瓶(方法1)或100 ml量瓶(方法2~6)中,用各自的溶出介质稀释至刻度,摇匀,滤过,弃去初滤液,取续滤液作为供试品溶液;另精密量取对照品储备液适量,用各自的溶出介质稀释制成每1 ml中含吲达帕胺5 μg(方法1)或2.5 μg(方法2~6)的溶液作为对照品溶液。精密量取供试品溶液和对照品溶液各20 μl,分别注入液相色谱仪,记录色谱图,计算各浓度水平的平均回收率,结果见表3。
2.2.4稳定性试验 取对照品溶液和供试品溶液分别在室温下放置0、2、8和24 h后进样测定峰面积,结果显示本品在24 h内稳定。
3 溶出度测定
3.1溶出度测定 取4批样品,按上述6种试验条件分别测定不同时间的累积溶出百分率,溶出曲线见图2。
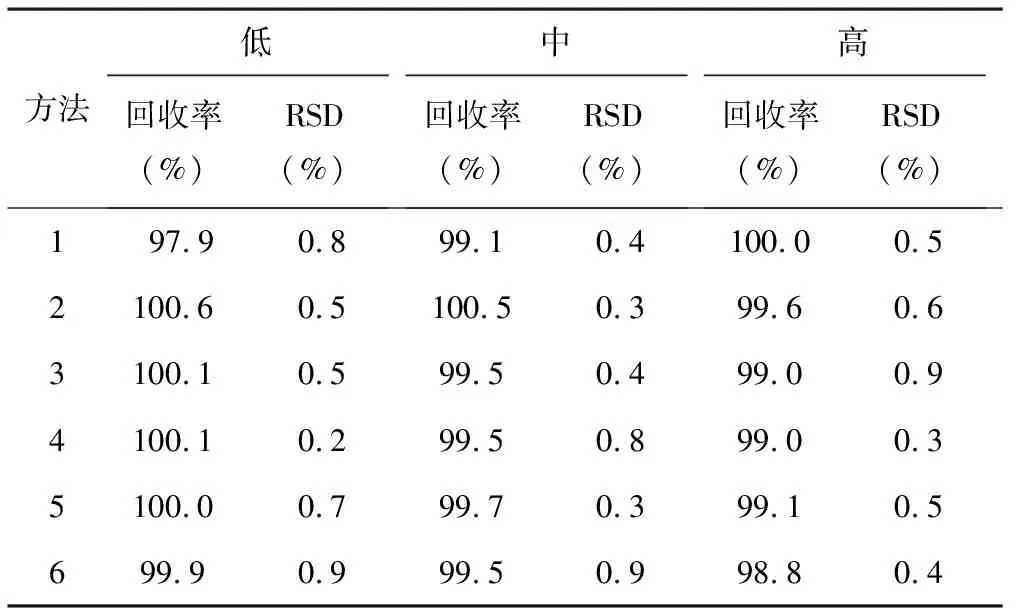
表3 回收率测定结果(n=3)


图2 6种测定条件下4批样品的溶出曲线
3.2数据处理及分析 采用美国FDA推荐使用的相似因子法。相似因子f2的数学表达式为:
式中:Ti为i时间参比制剂累积释药百分率;Ri为i时间受试制剂累积释药百分率;n为取样时间点个数。该方法计算的基本假设是受试制剂与参比制剂的累积溶出度差的平方和最小。
3.2.1在相同测定条件下国产制剂与进口制剂溶出行为的差异 以进口制剂(样品A)的溶出曲线为参比,分别将相同测定条件下国产样品的溶出曲线与其相比,计算f2值(表4)。以“相似因子f2≥42认为曲线相似”为判定原则,B厂制剂在上述试验条件下的溶出行为与进口制剂均相似;而C、D厂制剂无论溶出介质酸性如何,其溶出度与A制剂相比不相似。说明与进口制剂相比,国产制剂质量参差不齐,有些产品还有显著差异。

表4 与进口制剂(A)相比f2计算结果(n=4)
3.2.2不同测定条件下同批样品溶出行为的差异 以方法1中的溶出曲线为参比,每批样品在其他条件下的溶出曲线分别与其相比,计算f2值(表5)。结果表明,A、B、C三制剂溶出曲线间f2值均大于等于42,说明三家产品受溶出介质酸碱性、转速快慢等影响不大;而样品D在不同测定条件下f2值间有显著差异,说明其溶出行为受介质酸碱性影响很大,见图3。

表5 与方法1溶出曲线相比f2计算结果(n=4)

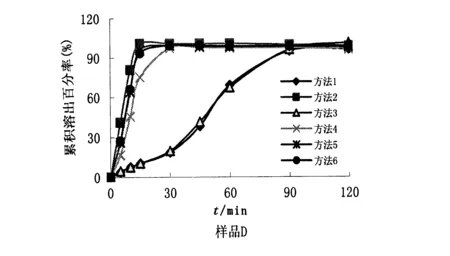
图3 4批样品在6种测定条件下的溶出曲线
4 讨论
4.1相似因子f2的计算过程中注意到比较时间点的选择,由于选取的点数过多或不合适都有可能导致差异较大的溶出曲线得出“相似”的错误结论,故每批样品选择重要、关键的溶出点位(n=4)进行比较,其中溶出量为85%以上的时间点最多选择一点。
4.2由于溶出度试验中所采用的溶出介质(主要是pH值不同的各种溶液)是模拟患者消化道的各种生理环境,转篮和桨板以及转速是模拟胃部和小肠的蠕动,因而,对于一个理想的药物来说,是应该在上述各种测定条件下都能具有稳定一致的溶出曲线,才有可能在任何年龄、任何体质患者中具有相似的溶出行为,进而产生同样的疗效。但从试验结果来看,国产制剂D在酸性介质中的溶出行为明显不同于其他pH介质,那么该药对体内胃酸水平不同的患者来说,极有可能产生不同的疗效,这在临床应用中具有指导意义,应该引起重视。
4.3从溶出曲线的测定结果不难看出,进口制剂具有令人满意的溶出曲线形状,且在各种试验条件下的溶出曲线几乎一致,显示了相当稳定的质量水平。国产B制剂溶出曲线与其相比较为相似,C、D制剂与其差异较大,说明国产制剂质量参差不齐,生产工艺、处方等尚需做深入探讨。另外,多家制剂体外溶出行为的这种差异是否与药物体内吸收切实相关,还需做进一步的体内试验研究。
1 中国药典.2005.二部.2005:253
2 中国药典.2010.二部.2010:345
3 谢沐风.简介日本“药品品质再评价”工程(溶出度研究系列一).中国药品标准,2005,6(6):42
猜你喜欢
中国药物经济学(2019年12期)2020-01-08
中成药(2018年1期)2018-02-02
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
特产研究(2016年3期)2016-04-12
中国卫生标准管理(2015年5期)2016-01-14
中国医药指南(2013年6期)2013-06-23
郑州大学学报(理学版)(2013年2期)2013-03-11
中国现代药物应用(2012年6期)2012-11-21
中国民族民间医药(2012年16期)2012-04-18
