
大鼠钙化血管中蛋白激酶G与钙调神经磷酸酶信号通路的交互调节作用
2011-06-09 03:39苑晓烨李绍冰李芳杨圣俊曾强
河北医药 2011年21期
苑晓烨 李绍冰 李芳 杨圣俊 曾强
动脉钙化在组织病理学上分为动脉内膜和动脉中层钙化。无论哪种钙化,都会引起血流动力学和机械力学的改变,最终将导致左心室肥大、心肌缺血和心律失常,并增加卒中和猝死的危险。血管钙化与骨代谢及血管平滑肌细胞(vascular smooth mucle cell,VSMC)表型转换关系密切。环三磷酸鸟苷依赖的蛋白激酶I(cGMP-dependent protein kinase,PKGI)及钙调神经磷酸酶(calcineurin,CaN)在参与调节血管平滑肌细胞增殖及骨代谢中发挥重要作用。既往研究发现培养VSMC内NO/PKG信号通路和/CaN信号存在交互调节作用[1]。但是,在血管钙化中NO-cGMP-PKG I通路与钙-CaN信号通路之间是否存在交互调节作用,进而影响血管钙化进程并不完全清楚。本研究旨在探讨大鼠血管钙化过程中NO-cGMP-PKG I通路与钙-CaN通路之间是否存在交互调节作用。
1 材料与方法
1.1 材料 华法令、维生素D3购自Sigma公司;环孢霉素A(CsA,Sandimmum)购自Novartis公司;维生素K1为无锡市第七制药有限公司生产;SD雄性大鼠购自河北医科大学实验动物中心,PKGI α-mRNA原位杂交检测试剂盒由武汉博士德生物工程有限公司合成,CaNA α mRNA原位杂交检测试剂盒由天津灏洋生物制品科技有限责任公司合成。
1.2 动物分组及钙化血管的制备 参照文献[2]将实验动物随机分为对照组、钙化组、钙化+CsA组(CsA组),每组12只。实验第1 ~8 天,3 组均给予维生素 K115 mg·kg-1·d-1,皮下注射,同时C组另给予CsA 10 mg·kg-1·d-1腹腔注射,A组和B组分别给予等量0.9%氯化钠溶液腹腔注射;实验第3天开始,B、C 组另给予维生素 D33 ×105U·kg-1·d-1,皮下注射,持续至实验第5天,及华法令15 mg/100 g,12 h 1次,皮下注射,持续至第8天,对照组在相同时间给予等量0.9%氯化钠溶液皮下注射;实验第9天,所有动物以10%水合氯醛腹腔注射麻醉后处死。
1.3 标本采集 大鼠处死前12 h禁食不限水,以10%水合氯醛腹腔注射麻醉后,将大鼠置于冰盒上,开胸,暴露出心脏,分离胸主动脉,并向下剖开腹部,分离出腹主动脉,从主动脉根部至腹主动脉分叉处剪下主动脉,迅速置于4℃预冷的0.9%氯化钠溶液中冲洗,直至盐水清凉,置于有0.1%二乙基焦碳酸酯(DEPC)的多聚甲醛溶液中,经常规梯度酒精脱水,二甲苯透明,石蜡包埋,每段血管均匀切片3~4张,分别做HE染色等形态学观察和免疫组化、原位杂交等检测。
1.4 HE染色 石蜡切片脱蜡至水;苏木素染色10 min;自来水冲洗2~5 min;1%的盐酸酒精分化;自来水冲洗;0.1%的氨水返蓝;自来水充分冲洗;0.5%伊红染色2 min;自来水冲洗;常规梯度酒精脱水、二甲苯透明,中性树胶封片。
1.5 原位杂交测定
1.5.1 PKGI α-mRNA 探针合成序列:①5’-AGACCTACAGGTCCTTCCACGACCTGCGCCAGGCG 3’;②5’-TGATCGACAATGTTTTCAAACAATAATGATGAGAA 3’;③5’-ACACGAGACAGCAGGAGCACATCCGCTCAGAGAAG 3’。步骤:石蜡切片脱蜡至水;置过氧化氢封闭液室温15 min;切片上滴加3%柠檬酸新鲜稀释的胃蛋白酶;滴加预杂交液;滴加杂交液覆盖组织44℃湿盒孵育过夜;杂交后的洗涤;滴加封闭液;滴加生物素化鼠抗地高辛;滴加SABC;滴加生物素化过氧化物酶,原位杂交用PBS洗;DAB显色,苏木素复染;常规梯度酒精脱水、二甲苯透明,中性树胶封片。
1.5.2 CaN Aα-mRNA 探针合成序列:①5’-CGGTG ACTTG GCGGA AATGG AACGG CTT;②5’-AAGAA GAGGT AGCGA GTGTT GGCAG GAG;③5’-CGAAG GCATC CATAC AGGCG TCATA AAC。探针①、②和③混合标记地高辛。步骤:石蜡切片脱蜡至水;置打孔液中室;置过氧化氢封闭液室温20 min,0.01 mol/L PBS冲洗3次;滴加复合消化工作液室,0.01 mol/L PBS冲洗3次,0.2×ssc(标准柠檬酸盐溶液)洗1次;滴加预杂交工作液覆盖组织37℃湿盒孵育1 h;预杂交后的洗涤;滴加杂交工作液覆盖组织42℃湿盒孵育过夜;杂交后的洗涤;滴加大鼠抗地高辛生物素标记的抗体工作液,覆盖组织37℃湿盒孵育45 min,0.01 mol/L PBS冲洗3次;滴加高敏过氧化物酶链亲和素复合物工作液;DAB显色,苏木素复染;常规梯度酒精脱水、二甲苯透明,中性树胶封片。
1.6 主动脉组织ALP活性测定 按照试剂盒说明书操作。定义每克组织蛋白(gprot)在37℃与基质作用15 min产生1 mg酚为1单位。计算公式为:组织ALP活性(U/gprot)=(测定管吸光度 OD值/标准管吸光度 OD值 ×标准管酚含量(0.003 mg)/取样量中的蛋白克数。
1.7 将原位杂交结果 应用Image-Pro Plus图象分析软件,分别计算各组主动脉组织相同面积内阳性细胞的累积光密度值(integrated optic density,IOD)。
1.8 统计学分析应用SPSS 13.0统计软件,计量资料以表示,对计量资料进行正态性检验及方差齐性检验,组间资料比较应用单因素方差分析,两两比较应用LSD-t检验,P<0.05为差异有统计学意义。
2 结果
2.1 HE染色 结果显示对照组大鼠主动脉壁结构完整,中层血管平滑肌细胞(VSMCs)排列整齐(图1);钙化组大鼠主动脉中层VSMCs排列紊乱,弹力纤维迂曲断裂(图2);CsA组大鼠主动脉中层VSMCs亦排列紊乱,弹力纤维层迂曲断裂(图3)。
2.2 原位杂交 3组大鼠主动脉可见 CaN Aα mRNA及PKGIα mRNA表达,主要位于胞浆中。见图4~6。钙化组与对照组比较:钙化组大鼠主动脉CaN Aα mRNA较对照组表达明显增加(P<0.01),PKGIα mRNA较对照组表达明显减少(P<0.01);CsA组与对照组比较:CsA组大鼠主动脉CaNAα mRNA较对照组表达明显增加(P<0.01),PKGIα mRNA较对照组表达明显增加(P<0.01);CsA组与钙化组比较:CsA组大鼠主动脉CaN Aα mRNA与钙化组比较表达明显减少(P<0.01),PKGIα mRNA 较钙化组表达明显增加(P<0.01)。见表1。
表1 原位杂交结果及主动脉组织ALP活性测定n=6,

表1 原位杂交结果及主动脉组织ALP活性测定n=6,
注:与对照组比较,*P<0.01;与钙化组比较,#P<0.05,△P<0.01
组别 CaN A αmRNA(IOD)PKGIα mRNA(IOD) ALP(U/gprot)对照组 10302±2636 8713±740 360±49钙化组 26421±1698* 5774±643* 463±65*CsA组 16775±4030# 11044±972# 480±75#
2.3 ALP活性 钙化组ALP活性(463±65)U/gprot较对照组(360±49)U/gprot增加(P<0.05);CsA组ALP活性(480±75)U/gprot较对照组(360±49)U/gprot明显增加(P<0.01),CsA组ALP活性(480±75)U/gprot与钙化组(463±65)U/gprot比较差异无统计学意义(P >0.05)。
3 讨论
以往认为血管钙化是衰老时出现的异位骨化,是血管疾病终末期所出现的变性表现,是钙盐在细胞内和细胞外基质的被动沉积。然而新近的许多研究发现,血管钙化并不是磷酸钙结晶在血管壁的简单而被动的沉积,而是钙盐沉积在细胞内(细胞内钙超载)和细胞外基质的复杂的、主动的、并且可能是高度可调的过程,类似于骨和软骨形成过程中的骨化。
本研究钙化血管中,大鼠主动脉PKGIα mRNA表达减少,CaN Aα mRNA表达增加,推测PKG影响CaN的表达。近年证实血管钙化是一个受调控的、类似于骨形成的主动过程,钙化的动脉有成骨细胞和破骨细胞,钙化的基质被骨基质取代,有新生血管侵入[3,4]。在骨组织,CaN、PKG 是破骨细胞发育必需的[5],有研究证实CaN、PKG还参与成骨细胞分化及成骨作用[6-8],CaN及PKG很可能通过成骨相关机制参与调节血管钙化。VSMCs能够向成骨细胞表型转化,是血管钙化的细胞学基础之一。研究证明CaN及PKG参与调节VSMCs分化,在血管成形术后再狭窄[9,10]及新生内膜形成中[11,12]具有重要作用。CaN、PKG很可能也参与VSMCs向成骨细胞转化和钙化。另外,血管钙化与 VSMCs凋亡密切相关[13,14]。CaN、PKG 参与 T淋巴细胞、心肌细胞等多种细胞的凋亡,CaN很可能参与VSMCs的凋亡。CaN信号通路在VSMC增殖及功能维持中起到重要作用。胞内Ca2+浓度控制着不同的细胞功能,包括基因表达、增殖、凋亡、粘附和迁移等,CaN可通过其去磷酸化作用调节Ca2+通道、影响胞内Ca2+浓度、参与VSMC增殖等功能活动的调控。一氧化氮(nitric oxide,NO)能够激发多种信号传导通路,刺激可溶性鸟苷环化酶催化细胞内cGMP合成,再由cGMP激活PKG调节局部和全身信号等。同时NO通过抑制肌浆网IP3受体和雷尼丁受体释放Ca2+,以及介导cGMP和PKG抑制N型钙通道Ca2+内流,调节平滑肌细胞内钙离子浓度。PKG是广泛存在于真核细胞内的一种丝(苏)氨酸蛋白激酶,存在于多种组织细胞,是重要的信号分子NO的下游底物,能够在多种水平调节Ca2+水平,从而参与多种细胞功能调节。Fiedler等[15]发现PKG激活后可以抑制Ca2+从L型钙通道进入胞质,可以抑制CaN-NAFT信号通路,从而减少心肌细胞体积。故推测,在血管钙化过程中,PKG可通过Ca2+的浓度变化影响CaN的表达。

图1 对照组大鼠主动脉HE染色(HE×400)
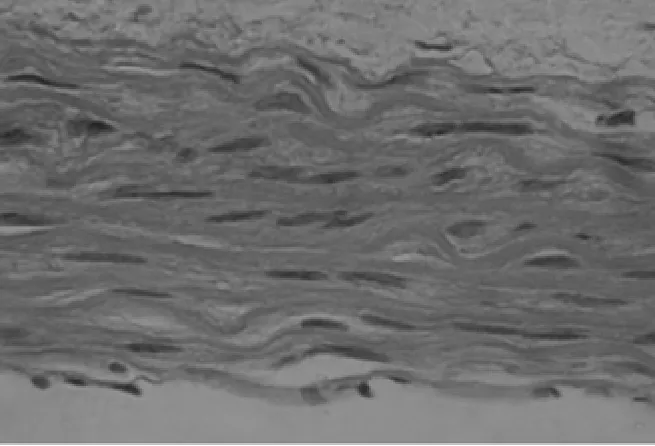
图2 钙化组大鼠主动脉HE染色(HE×400)
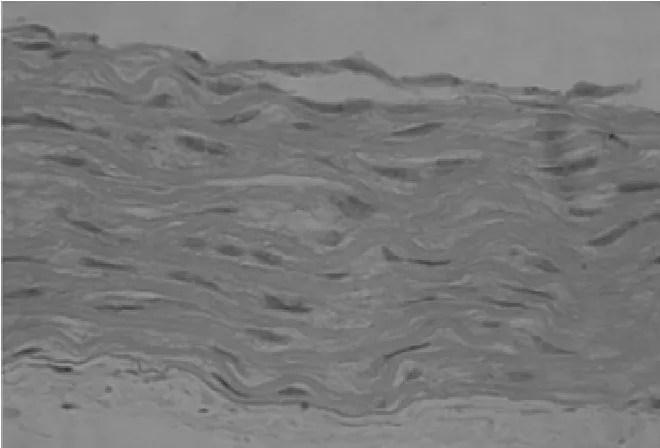
图3 CsA组大鼠主动脉HE染色(HE×400)
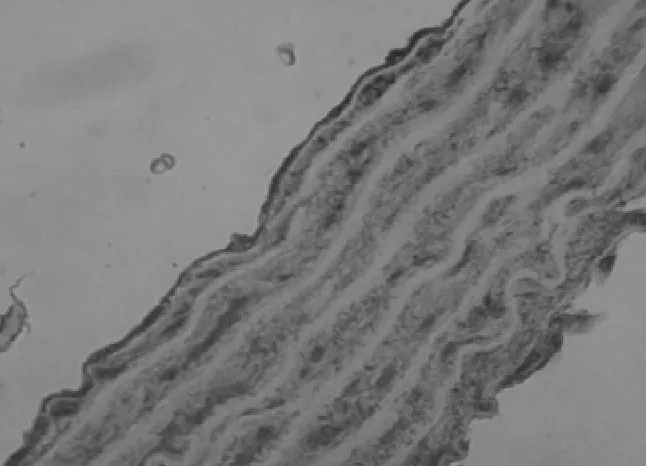
图4 对照组PKGⅠα mRNA表达(HE×400)
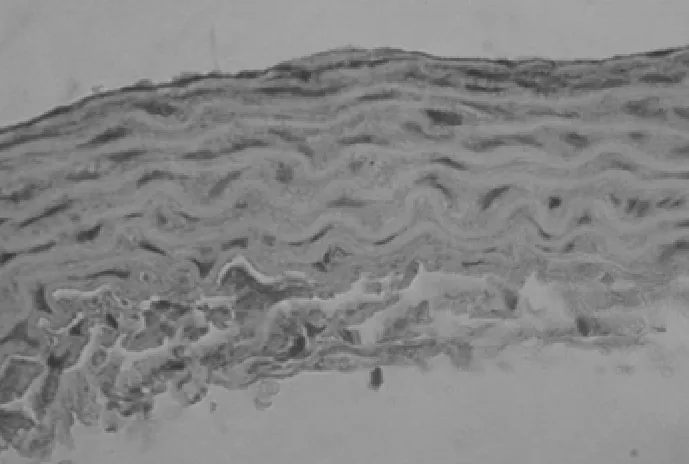
图5 钙化组PKGⅠα mRNA表达(HE×400)
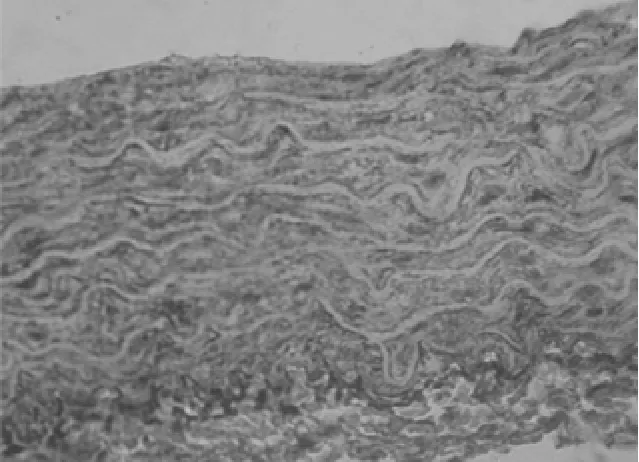
图6 CsA组PKGⅠα mRNA表达(HE×400)
本实验给予CaN抑制剂CsA后,钙化血管中CaN Aα mRNA表达减少,而PKG Iα mRNA的表达及钙化的标志物ALP却有增加的趋势,推测CaN抑制钙化血管中PKG Iα的表达。其机制可能为:CaN介导的脱磷酸化和核转位作用在信号传导途径中是一中枢性事件。它不仅本身可介导多条信号传导通路,而且通过其去磷酸化作用可对其他信号通路进行调节,使Ca2+信号与其他第二信使的调节机制发生“交谈”,协同调节细胞的功能。在钙化血管中CaN激活,使受磷蛋白(phospholamban,PLB)去磷酸化而活化,PLB能够抑制肌浆网钙-ATP酶(SERDA2a)的活性,从而减少肌浆网对胞浆内Ca2+的重吸收,使细胞浆内Ca2+水平升高,上调一氧化氮合酶活性,NO水平增加,继而生成大量cGMP,激活蛋白激酶A,抑制PKG Iα表达[16]。当用特异性抑制物CsA阻断CaN活性后,上述过程被抑制,从而使PKG Iα表达明显增加。
本研究结果提示,培养钙化血管内NO/PKG信号通路和Ca2+/CaN信号存在交互调节作用。血管钙化可以引起血流动力学和机械力学的改变,动脉管壁僵硬,收缩压升高、舒张压降低,脉压差增大,冠状动脉灌注降低,最终将导致左心室肥大、心肌缺血和心律失常,并增加卒中和猝死的危险[17,18]。已经证实CaN-NFAT信号通路是促进细胞肥大重要调节者,明确CaN与PKG I之间的相互关系有助于了解血管钙化的可能发病机制,并为抗血管钙化的药物治疗提供了可能的新途径。
1 孙宁玲,马旃,李世军.血管平滑肌细胞增殖中蛋白激酶G与钙调磷酸酶信号通路的交互调节.中国动脉硬化杂志,2006,14:645-648.
2 Price PA,Faus SA,Williamson MK.Warfarin-induced artery calcification is accelerated by growth and vitamin D.Arterioscler Thromb Vasc Biol,2000,20:317-327.
3 Johnson RC,Leopold JA,Loscalzo J.Vascular calcification pathobiological mechanisms and clinical implications.Circ Res,2006,99:1044-1059.
4 Shao JS,Cai J,Towler DA.Molecular mechanisms of vascular calcification lessons learned from the aorta.Arterioscler Thromb Vasc Biol,2006,26:1423-1430.
5 Sun L,Peng YZ,Zaidi N,et al.Evidence that calcineurin is required for the genesis of bone-resor-bing osteoclast.Am J Physiol Renal Physiol,2007,292:F285-F291.
6 Sun L,Blair HC,Peng Y,et al.Calcineurin regulates bone formation by the osteoblast.PNAS,2005,102:17130-17135.
7 Winslow MM,Pan M,Starbuck M,et al.Calcineurin/NFAT signaling in osteoblasts regulates bone mass.Dev Cell,2006,10:771-782.
8 Chikuda H,Kugimiya F,Hoshi K,et al.Cyclic GMP-dependent protein kinase II is a molecular switch from proliferation to hypertrophic differentiation of chondrocytes.Genes Dev,2004,18:2418-2429.
9 Yu HX,Sliedregt-bol K,Overkleeft H,et al.Therapeutic potential of a synthetic peptede inhibitor of nuclear factor of activated T cells as antirestenotic agent.Arterioscler Thromb Vasc Biol,2006,26:1531-1537.
10 Doi K,Ikeda T,Itoh H,et al.C-type natriuretic peptide induces redifferentiation of vascular smooth muscle cells with accelerated reendothelialization.Arterioscler Thromb Vasc Biol,2001,21:930-936.
11 Takeda R,Suzuki E,Takahashi M,et al.Calcineurin is critical for sodium-induced neointimal formation in normotensive and hypertensive rats.Am J Physiol Heart Circ Physiol,2008,294:H2871-H2878.
12 Sinnaeve P,Chiche JD,Gillijns H,et al.Overexpression of a constitutively active protein kinase G mutant reduces neointima formation and in-stent reste-nosis.Circulation,2002,105:2911-2916.
13 Shroff RC,Mcnair R,Figg N,et al.Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis.Circulation,2008,118:1748-1757.
14 Clarke MCH,Littlewood TD.Figg N,et al.Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration.Circ Res,2008,102:1529-1538.
15 Fiedler B,Lohmann SM,Smolenski A.Inhibition of calcineurin-NFAT hypertrophy signalingby cGMP-dependentproteinkinase type I in cardiac myocytes.PNAS,2002,99:11363-11368.
16 Pilz RB,Casteel DE.Regulation of gene expression by cyclic GMP.CircRes,2003,93:1034-1046.
17 Raggi P,Bellasi A.Clinical assessment of vascular calcification.Adv Chronic Kidney Dis,2007,14:37-43.
18 EI-Abbadi M,Giachelli CM.Mechanisms of vascular calcification.Adv Chronic Kidney Dis,2007,14:54-56.
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
昆明医科大学学报(2022年2期)2022-03-29
汽车维修与保养(2021年8期)2021-02-16
工业设计(2016年4期)2016-05-04
特产研究(2016年3期)2016-04-12
中国当代医药(2015年17期)2015-03-01
郑州大学学报(医学版)(2015年1期)2015-02-27
哈尔滨医药(2014年4期)2014-02-27
现代检验医学杂志(2014年4期)2014-02-02
中国医学科学院学报(2013年2期)2013-03-11
