
常见遗传性聋致病基因研究进展及基因诊断的临床应用
2011-06-05 14:36:25周学军欧阳小梅刘学忠
听力学及言语疾病杂志 2011年1期
周学军 欧阳小梅 刘学忠
·综述·
常见遗传性聋致病基因研究进展及基因诊断的临床应用
周学军1,2欧阳小梅1刘学忠1
耳聋的原因分为遗传因素和环境因素,也可以是遗传和环境因素共同作用的结果。遗传性聋中30%的患者同时合并有外耳畸形或其他器官系统疾病,称为综合征型聋(syndromic hearing impairment SHI),其余70%的患者为非综合征型聋(non-syndromic hearing impairment,NSHI)。根据遗传模式,遗传性聋又分为常染色体显性(DFNB)、隐性(DFNA)、性连锁和线粒体母系遗传性聋。在NSHI中,DFNB占80%,DFNA约占15%~20%,性连锁和线粒体遗传性聋约占1%[1]。迄今为止,已发现400多个伴有听觉障碍的综合征,鉴定出与之相关的遗传缺陷30多个,而在NSHI中,已定位了100多个致病位点(http://webh01.ua.ac.be/hhh/),克隆了70多个耳聋基因。近年来,遗传性聋致病的分子生物学研究发展迅速,有关耳聋基因及其功能方面的研究成果令人瞩目,本文就常见遗传性耳聋基因的结构功能、表型特点、人群分布的研究现状进行综述。
1 综合征型聋
综合征型聋在语前聋患者中占30%,但极少出现语后聋,其正确诊断对于患者及其家族成员的伴发器官或系统(如肾脏、眼部等)病变监控具有重要的意义。临床上有些综合征容易诊断,有些则需要对可疑个体进行系统的检查才能获得诊断,由于肾脏和眼部是综合征型聋最常伴发病变的器官,而且临床表现较为隐蔽,因而需要进行详细的专科检查。常见综合征型聋的突变基因及临床特点见表1。
2 非综合征型聋
GJB2基因突变所致非综合征型遗传性聋为双侧对称性语前聋,主要的听力曲线图为残余型、斜坡型和平坦型,极少数为U型,但没有以低频下降为主的上升型曲线。听力损伤程度变异较大,可从轻度到极重度,例如35delG纯合子突变患者,大多数出现极重度聋,但仍有部分患者为中度聋,极少数为轻度聋[6]。基因型和表型的关系研究表明,GJB2突变中,出现2个缺失突变的患者的耳聋程度较缺失/错义杂合突变的患者重,而2个错义突变的患者耳聋程度最轻[6,7]。据推测,GJB2基因突变的这些表型变化,很可能与修饰基因的作用有关,但迄今尚未得到证实[8]。
由于GJB2基因突变在遗传性聋中的高发生率,不同种群中均存在某一优势突变,而且其基因短小,只有2个外显子,编码226个氨基酸,因此,GJB2基因突变筛查已经成为耳聋分子检测的最基本项目。35del G突变筛查在欧美国家、167del T突变筛查在犹太人群以及235delC突变筛查在东亚国家都已经获得了很好的开展。Liu等[9]对118名中国非综合征型聋先证者检测GJB2基因,发现235delC突变的比例最高。Dai等[10]对中国3 004例NSHI患者进行GJB2基因突变筛查,鉴定出488例(16.3%)至少携带一个235delC突变位点,其中纯合突变233例(7.8%),杂合突变255例(8.5%)。
2.2 SLC26A4基因-DFNB4 SLC26A4基因又名PDS基因,含有21个外显子,编码跨膜蛋白pendrin。Pendrin功能主要与碘/氯离子的转运有关,并在甲状腺、内耳和肾脏中高表达。SLC26 A4基因突变可导致Pendred综合征(PS)和非综合征型聋(DFNB4),均可导致颞骨的发育畸形,包括大前庭水管综合征(large vestibular aqueduct syndrome,LVAS)和Mondini畸形。研究表明,Pendred综合征患者完全丧失碘/氯转运功能,而DFNB4患者碘/氯转运功能仍存在,但处于较低水平。
迄今为止,在感音神经性聋患者中已发现150多种SLC26A4突变类型(http://www.healthcare.uiowa.edu/labs/pendredandbor/slc Mutations.htm),其中绝大部分是错义突变,另外还有框移突变和剪接点突变。不同人种中突变的类型和发生频率有很大差异,但大部分的病例都为散发。Campbell等[11]认为,在白种人中,最常见的SLC26A4突变位点是L236P,其次为T416P和IVS8+1G>A;在西班牙裔中,最常见突变为Q514K[12];而在亚洲,H723R在韩国和日本为频发突变[13~15],在中国,由LVAS或LVAS伴Mondini畸形导致的听力障碍中最常见的SLC26 A4基因突变是IVS7-2 A>G[16~18]。有研究显示IVS7-2A>G突变为始祖效应[19]。
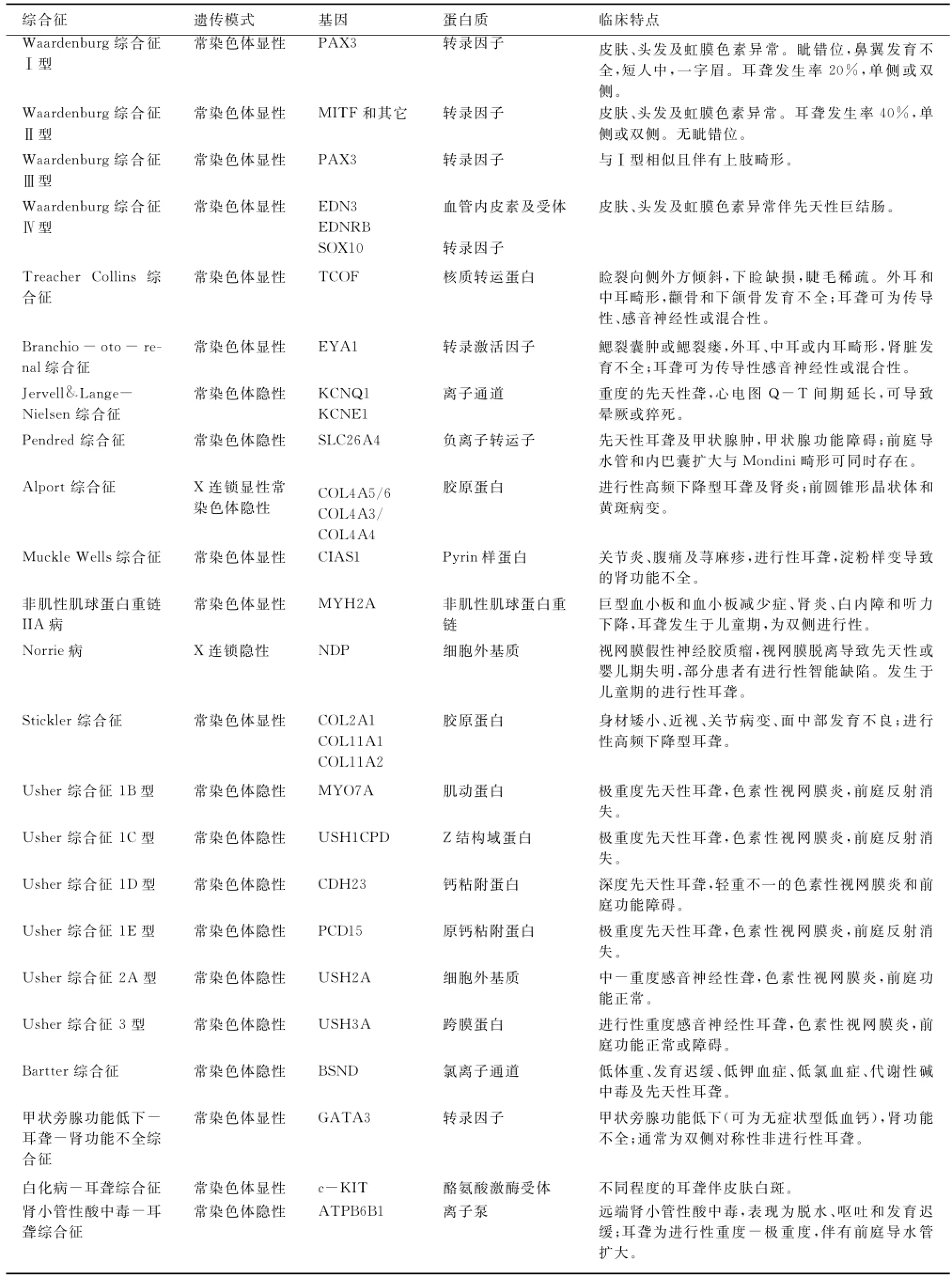
表1 常见综合征型聋
2.3 GJB6基因-DFNB1,DFNA3 GJB6基因位于染色体13q12上,编码连接蛋白CX30,与GJB2基因相邻。CX30与CX26在耳蜗内同一部位表达,并且两者编码的氨基酸序列约77%相同。其删除子从GJB6的5'prime末端延伸到GJB2基因,因此有可能GJB6的删除子也删除GJB2的区域。GJB6与GJB2共同构成了导致常染色体隐性感音神经性聋的突变位点DFNB1[20]。GJB2或GJB6的双等位基因突变或GJB2与GJB6共存的杂合突变均可导致NSHI。GJB6最常见的突变是342kb片段缺失,但随种群不同变异很大,发生率最高的是西班牙、法国、以色列和英国,占所有DFNB1等位基因突变的5.9%~9.7%,在所有GJB2单等位基因突变患者中,342kb片段缺失发生率达50%[21]。单倍型分析显示此突变在德系犹太人和西欧的一些国家中有明显的始祖效应存在[22]。另外2个GJB6突变位点是232kb和309kb片段缺失,不同种群中的发病率差异很大,中国目前尚无342kb、232kb和309kb片段缺失病例报道。
单个导致非综合征型显性遗传性聋的GJB6突变目前仅在少数患者报道。Grifa等通过对198名耳聋患者进行GJB6基因突变筛查,在一个显性遗传的意大利家系中发现T5M错义突变。与其他几个Connexin蛋白一样,GJB6基因的错义突变,还可以导致遗传性皮肤综合征(clouston syndrome)。
2.4 MYO7A基因-DFNB2,DFNA11 MYO7A基因定位于染色体11q13.5,有48个外显子,编码肌球蛋白myosinⅦA。myosinⅦA属于非常规肌球蛋白,在人胎儿内耳的内外毛细胞、前庭半规管的Ⅰ、Ⅱ型毛细胞均有表达,其功能目前尚不清楚,但认为其与细胞内膜的交通调控有关。MYO7A基因突变可导致DFNB2和DFNA11以及USH1B。目前已报告的突变位点在DFNB2有4个,DFNA11有5个,分别为在日本家系中发现的p.del A886-K887-K888、在美国家系中发现的p.G722R[23]、在荷兰家系中发现的p.N458I[24]、在德国家系中发现的p.R853C[25]和在意大利家系中发现的p.A230V[26];USH1B有126个(http://www.hgmd.cf.ac.uk/ac/gene.php?gene=MYO7A)突变分布于整个MYO7A基因。
北朝中后期,羁旅生涯、流亡生活、婚姻爱情等题材在表达方式上已经发生了潜在的变化,由直爽转变为委婉。如《紫翎马歌辞》:“高高山头树,风吹叶落去,一去数千里,何当还故处?”诗歌把动乱时期背井离乡的人,比作被山风吹落飘向远方的树叶,找不到自己的故里,委婉含蓄,给人很贴切的感觉。又如北魏胡太后所作《杨白花歌》:
DFNB2临床表现为非特异性的语前或语后聋,而DFNA11的表型特点为进行性语后聋,无或仅有轻微的前庭功能障碍,听力损失的程度、听力曲线图较为多变,与突变的位点有关,p.A230V、p.R853C和p.del A886-K887-K888的听力曲线为平坦型或高频下降型,p.N458I和p.G722R则为上升型曲线[23~26]。
2.5 GJB3基因-DFNA2 GJB3基因定位于人类染色体1p33-p35,编码有270个氨基酸的缝隙连接蛋白Connexin31。最早在中国的2个DFNA小家系中发现该基因的错义突变和无义突变,从而定位并成功克隆了GJB3基因。同时,动物实验证明GJB3基因在大鼠耳蜗中有表达。Liu等在25个中国DFNB家系中检测GJB3,发现其中2个家系成员携带GJB3复合杂合突变,其中之一为423-425del ATT的3bp缺失,另一为423A>G突变,其结果证明了GJB3突变不仅能导致常染色体显性非综合征型聋,也能导致常染色体隐性非综合征型聋。DFNA2临床多表现为高频听力受损的进行性语后聋。
2.6 WFS1基因-DFNA6/14/38 WFS1基因定位于4p16.1,编码跨膜结构蛋白wolframin,主要分布在内质网,可能参与蛋白质转运及调节钙离子的动态平衡,其在外周听觉系统的表达目前尚未清楚。WFS1基因突变可以引起常染色体显性感音神经性聋(DFNA6/14/38)和常染色体隐性Wolframin综合征。目前已经至少发现了该基因的30多种导致DFNA6/14/38的错义突变[27,28]和200多种导致Wolframin综合征的突变(http://www.khri.med.umich.edu/research/lesperance_lab/low_freq.php)。研究显示当WFS1基因发生失活突变时导致wolframin综合征,而在编码C-末端第8外显子的非失活性杂合错义突变则导致低频下降为主的感音神经性聋[29]。
DFNA6/14/38患者临床表现为发展缓慢、双侧对称的低频听力下降[30,31],此不同于DIAPHI基因突变引起的DFNA1。言语辨别率良好,如果仅在2 k Hz及以下频率受累时,常不自觉耳聋的存在,随着年龄增大,高频区听力也逐渐受累,听力曲线变为平坦型。Wolframin综合征表现为典型的发展迅速的高频听力下降,在人群中的杂合性携带率为0.3%~1%。因此,对于有阳性家族史的低频感音性聋患者进行WFS1基因的筛查是有价值的。
2.7 COCH基因-DFNA9 COCH基因定位于14q12-q13,它编码cochlin蛋白,在耳蜗基蜗螺旋缘和螺旋韧带的纤维细胞以及前庭迷路内的半规管壶腹脊感觉上皮的间质细胞内高表达。目前在10多个DFNA9家系中共发现了12种COCH基因突变-V66G、G88E、W117R、P51S、I109N、A119T、I109T、C542F、G87W、M512T、C542Y和V104del[32]。
DFNA9是目前发现的伴有前庭功能障碍的常染色体显性非综合征型聋之一,其表型在不同人群中较一致,大部分家系表现为40~60岁之间的进行性感音神经性聋[33],个别家系耳聋进展于20~40岁之间。初期为高频听力受损,继之所有频率受累,直至60~80岁发展为重度-极重度聋,部分患者出现类似于梅尼埃病的前庭功能障碍。DFNA9耳聋的外显率几乎为100%,而在某些家系,前庭功能障碍也表现为完全的外显率[33],一般伴有前庭功能障碍的患者听力下降比较严重。
2.8 母系遗传线粒体基因突变 线粒体DNA(mt DNA)是除核DNA外唯一存在于细胞内的遗传物质。共有37个编码基因,编码13种m RNAs、2种r RNA和22种t RNA。在有性生殖中,只有卵子才能将线粒体遗传给受精卵,因此,线粒体基因突变只能遗传自母系。
线粒体基因突变性耳聋在不同人群中发病率不同,有相当一部分的语后聋患者源自于线粒体基因突变[34],但很少引起语前聋。线粒体基因突变性耳聋分为综合征型和非综合征型耳聋,导致的综合征有MERRF综合征、MELAS综合征、Pearson综合征、Kearns-Sayre综合征和母系遗传性糖尿病-耳聋综合征。对耳蜗的影响为外毛细胞功能丧失,而耳蜗移植治疗有效表明耳蜗神经未受影响。
多个线粒体基因突变可导致非综合征型聋,其中最常见的是12Sr RNA基因上1555A>G的突变。此突变最早报告于一个大的阿拉伯-以色列家族,大部分受累患者表现为婴儿期发病的重度-极重度聋,部分为成年发病,而个别成员听力正常,继而发现该家系耳聋的表型可能与8号染色体上的一个未知的显性基因有关。目前,大量研究表明1555A>G突变与氨基糖苷类抗生素(AmAn)所致的药物性聋有关[34]。据分析,当线粒体DNA存在1555 A>G突变时,在AmAn存在的条件下,线粒体ATP产生不足,引起细胞内外Na+、K+、Ca+等离子浓度失衡,导致耳蜗和前庭细胞损伤或死亡。携带1555A>G突变的个体对AmAn特别敏感,即使小剂量使用也可能致聋,由于不同修饰基因的作用,1555 A>G突变的表型变化也非常大[35]。1555A>G突变率在西班牙人中为27%。在日本,无氨基糖苷类药物接触史的极重度耳聋个体中1555A>G突变携带率为1%。其他导致非综合征型聋的线粒体基因突变包括t RNAser(UCN)上的7445A>G、7472insC、7510T>C和7511T>C突变。此外,12Sr RNA的961位点突变和1095T>C突变可能与药物性聋和非综合征型聋有关[36~38]。在中国,由于Am An的大量应用,线粒体基因突变性耳聋发病率显著不同,Li等[36]对中国散发的氨基糖苷类抗生素致聋和非综合征型聋的儿童进行了线粒体12SrRNA突变的系统分析,发现在这一人群中48%为氨基糖苷类药物性聋,1555A>G突变占氨基糖苷类药物性聋和非综合征型聋的13%和2.9%,961位点突变分别占1.7%和4.4%。
3 常见遗传性聋致病基因的诊断意义及临床应用
目前,在大部分发达国家,新生儿听力筛查已常规化,经过听力筛查发现的重度-极重度聋儿童,即可进行耳聋基因筛查。众多非综合征型聋基因中,GJB2、SLC26A4突变在不同耳聋人群中均占有极高的比率,个别基因突变又与患者某些临床特征相关联,这些基因又存在着外显子不大、有突变热点或区域的特点,适合采用测序、限制性酶切、斑点杂交或基因扫描技术进行筛查,这使得临床上采用正确的基因筛查策略,在短期内完成耳聋遗传学诊断成为可行。
3.1 在中国,GJB2、SLC26A4和线粒体基因突变导致的耳聋占遗传性聋的30%~50%[39,40],因此,新生儿出生后即针对频发的GJB2、SLC26A4、线粒体1555A>G突变进行筛查,可以快速简便的早期发现相当一部分遗传性聋患儿。
3.2 耳聋如与氨基糖苷类抗生素使用相关,或者有明确的母系遗传特征,须进行线粒体基因突变筛查,其中12SrRNA基因上的1555A>G突变为筛查重点。对携带mt DNA 1555A>G突变人群明确告知终生禁用氨基糖苷类药物,可有效避免药物性聋的发生。
3.3 SLC26A4基因突变发病率在ARNSHI中仅次于GJB2突变,临床上表现为进行性的高频下降型耳聋或同时伴有甲状腺肿、前庭导水管扩大或Mondini畸形的患者,须重点筛查SLC26A4基因,筛查位点在欧美国家主要为L236P、T416P、IVS8+1G>A,在亚洲则主要为IVS7-2A>G和H723R。在大前庭水管综合征患者,鉴定出SLC26A4基因突变后,相当一部分尚无听力障碍或仍具有较好的残余听力,对此类患者早期诊断,可以为其制订一整套的生活指导方案,预防耳聋的发生或进一步加重。由于SLC26A4基因突变筛查能够先于CT检查之前发现和确诊LVAS,而且检出率大于80%,在某些地区更容易进行批量检测,因而已经成为颞骨CT筛查的重要补充方法。
3.4 临床表现为发展缓慢的、双侧对称的2 k Hz以下低频听力下降,须考虑筛查WFS1基因和MYO7A基因。
3.5 临床表现为迟发的进行性听力下降伴进行性前庭功能障碍患者,提示重点筛查COCH基因。
3.6 听力筛查中出现ABR反应阴性而OAE反应阳性的儿童,提示筛查OTOF基因。
3.7 耳聋遗传模式为X-连锁且伴有Mondini畸形等骨迷路的发育不良患者,应考虑筛查POU3F4基因。如X-连锁中-重度进行性感音神经性聋,应考虑筛查PRPS1基因[41]。
耳聋基因诊断明确后,根据先证者及其父母亲携带基因类型,结合遗传规律,即可提供准确的遗传咨询,评估再生育聋儿的风险进行耳聋产前诊断和耳聋基因远程诊断。
1 Cryns K,Van Camp G.Deafness genes and their diagnostic applications[J].Audiol Neurootol,2004,9:2.
2 Kenneson A,Van Naarden Braun K,Boyle C.GJB2(connexin 26)variants and nonsyndromic Sensorineural hearing loss:A Huge review[J].Genet Med,2002,4:258.
3 Van Laer L,Coucke P,Mueller RF,et al.A common founder for the 35delG GJB2 gene mutation in connexin 26 hearing impairment[J].J Med Genet,2001,38:515.
4 Ohtsuka A,Yuge I,Kimura S,et al.GJB2 deafness gene shows a specific spectrum of mutations in Japan,including a frequent founder mutation[J].Hum Genet,2003,112:329.
5 Yan D,Park HJ,Ouyang XM,et al.Evidence of a founder effect for the 235delC mutation of GJB2(connexin 26)in east Asians[J].Hum Genet,2003,114:44.
6 Snoeckx RL,Huygen PLM,Feldmann D,et al.GJB2 mutations and degree of hearing loss:A multicenter study[J].Am J Hum Genet,2005,77:945.
7 Cryns K,Orzan E,Murgia A,et al.A genotype-phenotype correlation for GJB2(connexin 26)deafness[J].J Med Genet,2004,41:147.
8 Hilgert N,Huentelman MJ,Thorburn AQ,et al.Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene[J].Eur J Hum Genet,2009,17:517.
9 Liu XZ,Xia XJ,Ke XM,et al.The prevalence of connexin 26(GJB2)mutations in the Chinese population[J].Hum Genet,2002,111:394.
10 Dai P,Yu F,Han B,et al.The prevalence of the 235delC GJB2 Mutation in a Chinese deaf population[J].Genet Med,2007,9:283.
11 Campbell C,Cucci RA,Prasad S,et al.Pendred syndrome,DFNB4,and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations[J].Hum Mutat,2001,17:403.
12 Pera A,Dossena S,Rodighiero S,et al.Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA[J].Procn Natl Acad Sci USA,2008,105:18 608.
13 Park HJ,Shaukat S,Liu XZ,et al.Origins and frequencies of SLC26A4(PDS)mutations in east and south Asians:Global implications for the epidemiology of deafness[J].J Med Genet,2003,40:242.
14 Park HJ,Lee SJ,Jin HS,et al.Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans[J].Clin Genet,2005,67:160.
15 Tsukamoto K,Suzuki H,Harada D,et al.Distribution and frequencies of PDS(SLC26A4)mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct:A unique spectrum of mutations in Japanese[J].Eur J Hum Genet,2003,11:916.
16 Reyes S,Wang G,Ouyang X,et al.Mutation analysis of SLC26A4 in mainland Chinese patients with enlarged vestibular aqueduct[J].Otolaryngol Head Neck Surg,2009,141:502.
17 Wang QJ,Zhao YL,Rao SQ,et al.A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China[J].Clin.Genet,2007,72:245.
18 Dai P,Li Q,Huang D,et al.SLC26A4 c.919-2A4G varies among Chinese ethnic groups as a cause of hearing loss[J].Genet Med,2008,10:586.
19 Yang JJ,Tsai CC,Hsu H M,et al.Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutationin the PDS gene[J].Hear Res,2005,199:22.
20 Common JE,Bitner-Glindzicz M,O'Toole EA,et al.Specific loss of connexin 26 expression in ductal sweat gland epithelium associated with the deletion mutation del(GJB6-D13S1830[J].Clin Exp Dermatol,2005,30:688.
21 Del Castillo I.A deletion involving the connexin 30 gene in nonsyndromic hearing impairment[J].N Engl J Med,2002,346:243.
22 Del Castillo I,Moreno-Pelayo MA,Del Castillo FJ,et al.Prevalence and evolutionary origins of the del(GJB6-D13S1830)mutation in the DFNB1 locus in hearingimpaired subjects:A multicenter study[J].Am J Hum Genet,2003,73:1 452.
23 Street VA,Kallman JC,Kiemele KL.Modifier controls severity of a novel dominant low frequency Myosin VIIA(MYO7A)auditory mutation[J].J Med Genet,2004,41:e62.
24 Luijendijk MW,Van Wijk E,Bischoff AM,et al.Identification and molecular modelling of a mutation in the motor head domain of myosin VIIA in a family with autosomal dominant hearing impairment(DFNA11)[J].Hum Genet,2004,115:149.
25 Bolz H,Bolz SS,Schade G,et al.Impaired calmodulin binding of myosin-7A causes autosomal dominant hearing loss(DFNA11)[J].Hum Mutat,2004,24:274.
26 Di Leva F,D'Adamo P,Cubellis MV,et al.Identification of a novel mutation in the myosin VIIA motor domain in a family with autosomal dominant hearing loss(DFNA11)[J].Audiol Neurootol,2006,11:157.
27 Fukuoka H,Kanda Y,Ohta S,et al.Mutations in the WFS1 gene are a frequent ause of autosomal dominant nonsyndromic low-frequency hearing loss in Japanese[J].Jhum Genet, 2007,52:510.
28 Khanim F,Kirk J,Latif F,et al.WFS1/wolframin mutations,Wolfram syndrome,and associated diseases[J].Hum Mutat,2001,17:357.
29 Cryns K,Pfister M,Pennings RJ,et al.Mutations in the WFS1 gene that cause low frequency sensorineural hearing loss are small non-inactivating mutations[J].Hum Genet,2002,110:389.
30 Young TL,Ives E,Lynch E,et al.Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1[J].Hum Mol Genet,2001,10:2 509.
31 Bespalova IN,Van Camp G,Bom SJ,et al.Mutations in the Wolfram syndrome 1 gene(WFS1)are a common cause of low frequency sensorineural hearing loss[J].Hum Mol Genet,2001,10:2501.
32 Hildebrand MS,Tack D,Deluca A,et al.Mutation in the COCH gene is associated with superior semicircular canal dehiscence[J].Am J Med Genet A,2009,149:280.
33 Kamarinos M,McGill J,Lynch M,et al.Identification of a novel COCH mutation,I109N,highlights the similar clinical features observed in DFNA9 families[J].Hum Mutat,2001,17:351.
34 Hutchin TP,Thompson KR,Parker M,et al.Prevalence of mitochondrial DNA mutations in childhood/congenital onset non-syndromal sensorineural hearing impairment[J].J Med Genet,2001,38:229.
35 Kokotas H,Petersen MB,Willems PJ.Mitochondrial deafness[J].Clin Genet,2007,71:379.
36 Li R,Greinwald JH,Yang L,et al.Molecular analysis of mitochondrial 12S rRNA and t RNAser(UCN)gene in paediatric subjects with nonsyndromic hearing loss[J].J Med Genet,2004,41:615.
37 Li Z,Li R,Chen J,et al.Mutational analysis of the mitochondrial 12S r RNA gene in Chinese paediatric subjects with aminoglycoside-induced and non-syndromic hearing loss[J].Hum Genet,2005,117:9.
38 Wang Q,Li R,Zhao H,et al.Clinical and molecular characterization of a Chinese patient with auditory neuropathy associated with mitochondrial 12S r RNA T1095C mutation[J].Am J Med Genet A,2005,133:27.
39 戴朴,于飞,康东洋,等.线粒体基因1555位点和GJB21基因及SLC26A4基因的诊断方法及临床应用[J].中华耳鼻咽喉头颈外科杂志,2005,40:769.
40 戴朴,刘新,于飞,等.18个省市聋校学生非综合征型聋病分子流行病学研究(1)-GJB2 235delC和线粒体DNA12Sr RNA A1555G突变筛查报告[J].中华耳科学杂志2006,4:1.
41 Liu XZ,Han DY,Li JZ,et al.Loss-of-Function Mutations in the PRPS1 gene cause non-syndromic X-linked sensorineural deafness,DFN2[J].Am J Hum Genet,2010,86:65.
(2009-12-07收稿)
(本文编辑 周涛)
10.3969/j.issn.1006-7299.2011.01.024
R764.44
A
1006-7299(2011)01-0073-05
1 迈阿密大学医学院耳鼻咽喉科(佛罗里达 33136); 2 海南医学院附属医院耳鼻咽喉科
刘学忠(Email:xliu@med.miami.edu)
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国现代医生(2022年19期)2022-11-04 10:13:29
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2021年8期)2021-11-02 06:24:54
中国民间疗法(2021年8期)2021-07-22 05:53:42
中国生殖健康(2020年4期)2021-01-18 02:58:32
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国生殖健康(2018年4期)2018-11-06 07:12:36
小学生导刊(2018年13期)2018-06-29 03:49:00
西南军医(2015年1期)2015-01-22 09:08:34
