
高效液相色谱法测定尼可刹米注射液的含量
2011-06-05 02:33姜锋卫河南省焦作市药品检验所河南焦作454000
中国医药科学 2011年21期
姜锋卫河南省焦作市药品检验所,河南焦作 454000
尼可刹米注射液为中枢兴奋药,临床上用于中枢性呼吸抑制及各种原因引起的呼吸抑制。为《中国药典》(2010年版)二部所收载,检测方法为分光光度法[1],文献报道的分析方法有GC[2]、磷光分析[3]。本研究用高效液相色谱法测定注射液中尼可刹米的含量,为控制药品质量,提供一种简单、灵敏、快速准确的检测方法。
1 仪器与试药
Agilent 1200高效液相色谱仪(包括G1322A在线脱气机、G1311A四元泵、G1329A进样器G1314B VWD检测器);92SM-202A电子天平。尼可刹米原料(天津药业焦作有限公司提供,含量为99.3%);尼可刹米注射液,规格1.5 mL∶0.375 g(北京市永康药业有限公司,10060224;天津药业焦作有限公司,批号10052322、10071922);色谱纯乙腈,纯净水。
2 方法与结果
2.1 色谱条件
色谱柱为Eclipse DCB-C18(150 mm×4.6 mm,5μm);流动相[4-5]为乙腈-水(20∶80);检测波长263 nm,流速1.0 mL/min,柱温30℃,进样量10μL。
2.2 对照品溶液
用天平称取尼可刹米原料0.553 g,置100 mL容量瓶中,加水溶解并稀释释至刻度,摇匀;得对照品溶液(5.490 mg/mL)。
2.3 供试品溶液的制备
取供试品5支,混匀。精密量取2 mL,置200 mL容量瓶中,加水稀释至刻度,摇匀,即得供试品溶液(2.5 mg/mL)。
2.4 空白对照试验
依据该药的处方取辅料适量,按供试品溶液制备方法制备不含尼可刹米的空白对照溶液。分别取对照品溶液10μL、供试品溶液20μL、空白对照溶液10μL进样。
从色谱图上看出,在该色谱条件下,供试品溶液和对照品溶液中均出现相应的尼可刹米色谱峰,而空白对照溶液未见色谱峰,表明空白样品不干扰测定。结果见图1。

图1 尼可刹米的色谱图
2.5 线性关系考察
精密量取浓度为5.490 mg/mL的对照液10 mL置100 mL容量瓶中,加水稀释至刻度,摇匀,得浓度为0.549 mg/mL的对照液,分别取浓度为0.553 mg/mL的对照液10μL,20μL,30μL;浓度为 5.490 mg/mL的对照液5μL,10μL,20μL注入液相色谱仪,测定峰面积。以峰面积(A)为纵坐标,浓度(C)为横坐标,进行线性回归,得回归方程:A=3595.7C-8.6,r=0.9993;表明在浓度为0.549~10.980 mg/mL的范围内进样,样品量与峰面积呈良好的线性关系。
2.6 精密度试验
取供试品溶液(2.5 mg/mL),按上述色谱条件重复进样6次,每次进样10μL,测定峰面积,RSD=0.8%(n=6)。
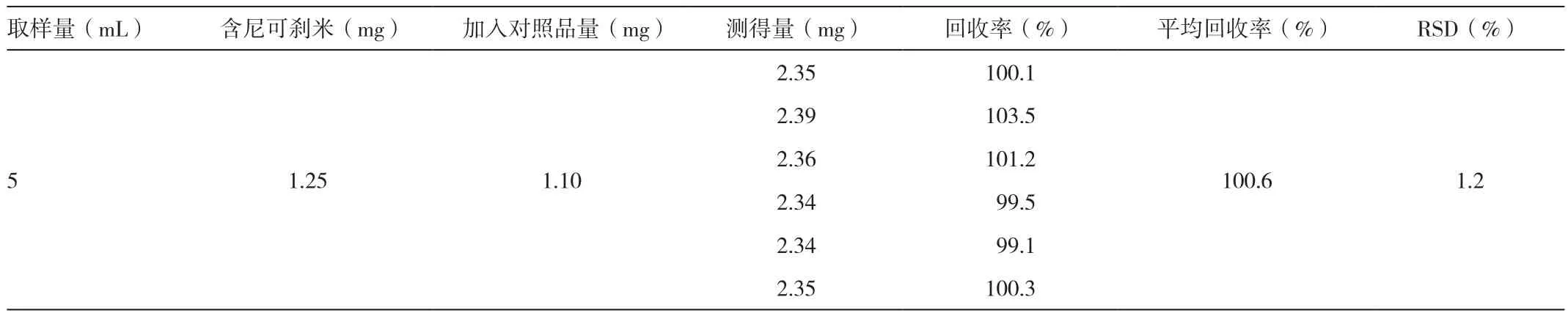
表1 回收率实验结果(n=6)
2.7 重复性试验
取同一批号样品,照供试品溶液制备方法制作供试品溶液6份,分别测定,测得供试品中尼可刹米的平均含量为100.3%,RSD=1.3%(n=6),表明方法的重复性良好。
2.8 稳定性试验
取供试品溶液,分别于 0,3,6,9,12 h 进样 10 μL,测定峰面积,结果RSD为0.95%(n=5),说明供试品溶液至少在12 h内稳定。
2.9 加样回收率试验
精密量取供试品溶液(2.5 mg/mL)5 mL,置于10 mL容量瓶中;精密量取对照品溶液(5.49 mg/mL)2 mL,加入上述容量瓶中,加水至刻度,混匀。依此法平行制备6份,依次进样,测定含量,结果见表1。
2.10 样品含量测定
分别精取对照品溶液和供试品溶液各10μL注入色谱仪,按外标峰面积法计算尼可刹米含量。见表2。

表2 样品含量测定结果(n=3)
3 讨论
取尼可刹米原料适量用流动相溶解,在200~400 nm的波长范围内进行扫描。结果在263 nm处有最大吸收,故选用263 nm为本方法的检测波长。
作者试验过程中,曾比较用乙腈-磷酸盐流动相和乙腈-水流动相,结果显示乙腈-水系统完全可以满足本实验的要求,主峰的峰形较好,考虑到减化操作,故采用乙腈-水系统。
分别用药典方法和本实验方法测定上述10052322、10060224、10071922三个样品,结果药典法分别为99.8%、96.9%、97.0%;本方法分别为100.3%、97.1%、96.9%,显示本方法与药典方法无明显差异,可用于尼可刹米注射液的含量测定。
[1] 国家药典委员会.中华人民共和国药典(二部)[S].2010:224.
[2] 候一斌.尼可刹米及其代谢产物的气相色谱/质谱法检测[J].质谱学报,1992,1(13):57-60.
[3] 黄如衡,周东,何长清.尼可刹米经犬气道给药后的药物动力学与药效学[J].中国药理学报,1994,15(3):271.
[4] 徐帆,冯恩富,徐贵丽,等.氨茶碱、多巴胺、尼可刹米注射液配伍稳定性研究 [J].中国药业,2009,18(23):12-14.
[5] 郑希林,吴瑞良.尼可刹米注射液的稳定性考查[J].中国药师,2000,3(6):380.
猜你喜欢
环境技术(2021年6期)2022-01-18
中国应急管理科学(2021年4期)2021-04-13
中国动物保健(2020年5期)2020-07-11
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
沈阳工程学院学报(自然科学版)(2019年3期)2019-07-26
考试周刊(2018年68期)2018-09-17
人民周刊(2016年11期)2016-06-30
中国猪业(2016年1期)2016-01-28
中国中医药现代远程教育(2014年11期)2014-08-08
