
甘草总皂苷的提取纯化工艺研究
2011-05-26 01:13:26朱中佳熊富良张雪琼冯井庆高文天
中成药 2011年2期
朱中佳, 熊富良, 张雪琼, 冯井庆, 高文天
(武汉理工大学化学工程学院,湖北武汉 430070)
甘草为豆科植物甘草Glycyrrhiza uralensisFisch、胀果甘草Glycyrrhiza inflataBat或光果甘草Glycyrrhiza glabraL.的干燥根及根茎,具有补脾益气,清热解毒,祛痰止咳,缓急止痛,调和诸药的功能[1]。甘草的主要成分为甘草酸等三萜皂苷和甘草苷为主的甘草黄酮。甘草中三萜类皂苷具有含量高、生理活性强的特点,不仅具有抗溃疡、消炎、解毒等生理活性,近年研究还发现具有抗病毒、抗癌活性[2]。而日本采用甘草总皂苷治疗慢性乙肝已有20多年的历史[3]。因此探讨甘草的提取和纯化工艺具有十分重要的意义。甘草中有效成分的提取方法包括水提取法[4]、索氏提取法[5]、超高压法[6]、微波辅助提取法[7]、超声波提取法[8]和超临界 CO2萃取法[9]等。而正交试验设计在优化甘草提取工艺中应用广泛[10-11]。甘草中有效成分的纯化方法则主要为大孔吸附树脂法[12-13]。本试验采用水提取法提取甘草总皂苷并采用正交试验法对其工艺进行了优选,接着以HPD系列的大孔树脂纯化了甘草总皂苷。
1 仪器与材料
Agilent 1100型高效液相色谱仪;UV1101型紫外分光光度仪,JA2003N型分析天平,AS5150A型超声波清洗器,DHG-9140A型电热恒温鼓风干燥箱。
HPD-100、HPD-300、HPD-700、HPD-722 和 D-101 型大孔吸附树脂由河北沧州宝恩化工有限公司提供。甘草酸单铵盐对照品(中国药品生物制品检定所),甘草购自武汉市神草中药饮片有限责任公司,由武汉理工大学化工学院刘小平教授鉴定。所用试剂均为分析纯。
2 方法与结果
2.1 分析方法
2.1.1 甘草中甘草酸含量测定方法(HPLC)
色谱条件:Kromasil C18(4.6 mm ×250 mm;5 μm);检测波长:250 nm;流速:1 mL/min;柱温:25℃;流动相为甲醇-0.2 mol/L醋酸铵溶液-冰醋酸(67∶33∶1)。
对照品溶液的制备:取甘草酸单铵盐对照品10 mg,精密称定,置50 mL量瓶中,用流动相溶解并稀释至刻度,摇匀,即得(每1 mL含甘草酸单铵盐对照品0.2 mg,折合甘草酸为0.195 9 mg)。
供试品溶液的制备:精密吸取0.1 g生药/mL的甘草提取液1 mL,加水稀释并定容至50 mL,摇匀,滤过,即得。
线性关系考察:精密吸取上述对照品溶液 1,2,4,6,8 mL,置10 mL量瓶中加流动相稀释至刻度。按上述的色谱条件,分别进样20 μL,测定其峰面积,并以对照品浓度为横坐标,峰面积为纵坐标作图,绘制标准曲线,得回归方程:Y=15 974X+100.4,r2=0.999 7。结果表明,甘草酸在0.019 6~0.156 8 mg/mL的浓度范围内呈良好的线性关系。
2.1.2 甘草总皂苷含量测定方法(UV)
对照品溶液的制备:精密称取甘草酸单铵盐对照品10.1 mg,置25 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,精密量取1 mL用甲醇稀释至10 mL,即得。
供试品溶液的制备:精密吸取样品溶液1 mL,加甲醇稀释并定容至50 mL,摇匀,滤过,即得。
最大吸收波长的确定:取对照品溶液,在紫外分光光度计上200~400 nm范围内进行扫描。结果表明在258 nm处有最大吸收,故选用258 nm作为测定波长。
线性关系考察:精密量取对照品溶液(40.4 μg/mL)1,2,4,6,8 mL,置 10 mL 量瓶中,加甲醇至刻度,258 nm 测定吸光度。以吸光度为纵坐标,浓度为横坐标,绘制标准曲线,得回归方程:Y=0.026 3X+0.013,r2=0.999 6,线性范围:4.04 ~32.32 μg/mL。
2.2 甘草总皂苷的提取工艺
通过预实验确定水为提取溶剂后,为优化水煎煮的条件,选用正交试验法,选定加水量、煎煮时间、煎煮次数作为考察的3个因素,每个因素各取3个水平(见表1)。采用L9(34)正交进行试验,以甘草酸和甘草总皂苷的提取率为考察指标,对结果进行方差分析,确定最佳提取工艺,见表2~4。
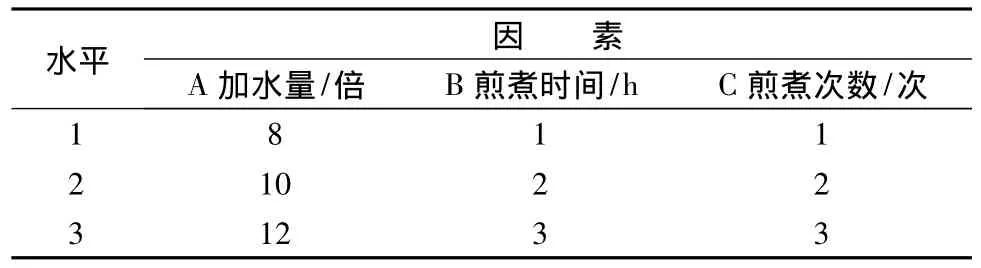
表1 因素水平表
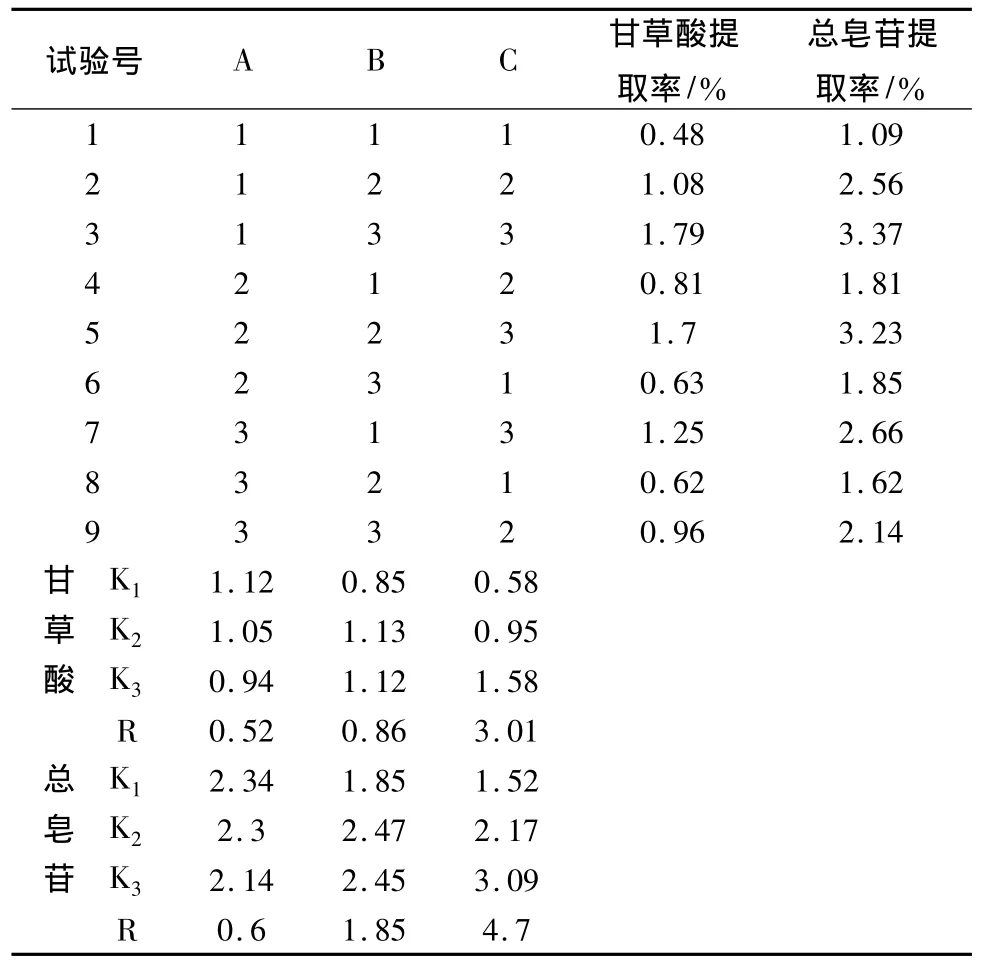
表2 正交试验
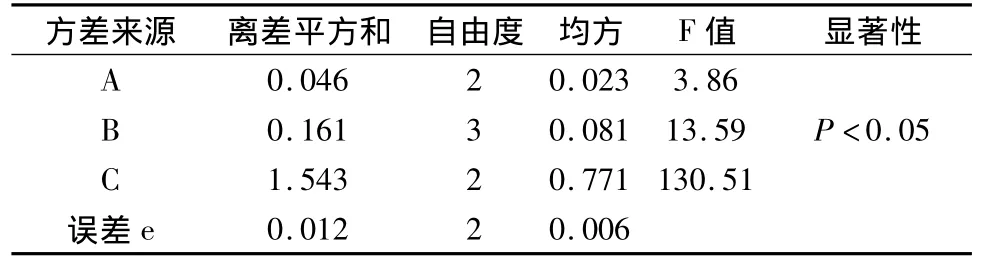
表3 方差分析(甘草酸)
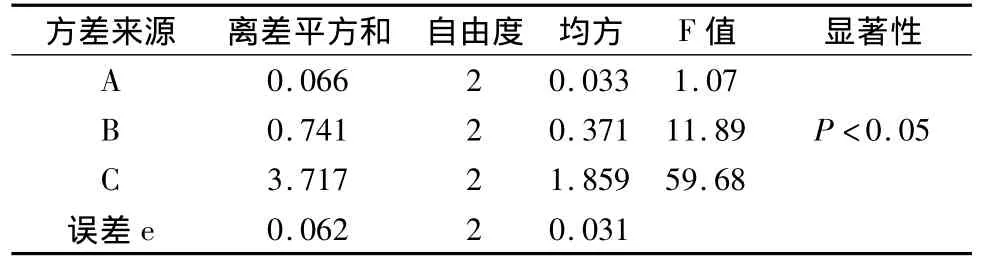
表4 方差分析(总皂苷)
由正交试验结果确定最佳提取工艺:加水量8倍,煎煮时间2 h,煎煮次数3次。按该提取工艺进行3次验证试验,结果甘草酸平均提取率为2.05%(RSD <2%),甘草总皂苷平均提取率为4.2%(RSD <2%)。以最佳提取工艺提取,提取液加水定容到含生药0.2 g/mL的溶液(甘草总皂苷含量为8.39 mg/mL),作为下一步实验上样液。
甘草酸提取率=所测甘草酸量(g)/所用甘草量(g)×100%
甘草总皂苷提取率=所测总皂苷量(g)/所用甘草量(g)×100%
2.3 大孔树脂纯化工艺
2.3.1 树脂型号筛选:精密称取上述5种大孔树脂(均相当于干树脂1 g),分装入50 mL具塞锥形瓶中,精密加入上样液50 mL(相当于0.2 g生药/mL),每30 min振摇1次,持续3 h后,静置24 h,上样液滤过,按2.1项下方法测定吸光度后,计算吸附量。滤出的各树脂另置于50 mL具塞锥形瓶中,精密加入70%乙醇50 mL,每30 min振摇1次,持续3 h,静置24 h后滤过,测定吸光度,计算解吸率,结果见表5。综合分析,HPD-300树脂为甘草总皂苷纯化的最佳树脂。另取3份HPD-300树脂(均相当于干树脂1 g),按上述方法进行3次平行实验,结果平均比吸附量170.14 mg/g(RSD<2%),平均解吸率87.65%(RSD<2%),故选取 HPD-300树脂为甘草总皂苷纯化所用树脂。
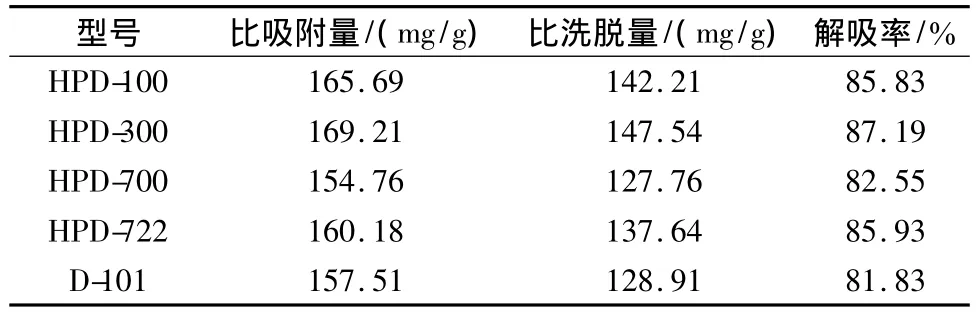
表5 大孔树脂对甘草总皂苷的吸附及解吸的影响
比吸附量=(上样液中总皂苷的质量浓度×体积-吸附后溶液中总皂苷的质量浓度×体积)/干树脂质量
比洗脱量=洗脱液中总皂苷的质量/干树脂质量
解吸率=(洗脱液质量浓度×体积)/饱和吸附量×100%
2.3.2 上样液质量浓度的确定:准确称取处理好的HPD-300型树脂3份(均相当于干树脂4 g),湿法装柱。取上样液(生药0.2 g/mL)50 mL,分别加水稀释0、1、2倍,制备成质量浓度分别为0.2 g/mL、0.1 g/mL、0.05 g/mL的上样液,每个质量浓度上样液平行上柱,以2 mL/min的流速吸附完全后,先以4 BV水冲洗,再换用4 BV 70%乙醇洗脱,收集洗脱液,得液体样品。按2.1项下方法测定吸光度后,计算洗脱液中总皂苷比洗脱量,结果分别为75.6 mg/g、69.4 mg/g、67.5 mg/g。可见,在上柱液中总皂苷质量固定的情况下,上柱液质量浓度为0.2 g生药/mL时洗脱效果最好。
2.3.3 最大上样量的确定:准确称取处理好的HPD-300树脂装柱(相当于干树脂4 g),精密吸取上样液200 mL,以流速2 mL/min通过树脂柱,每1BV接收一次流出液,并测定每份流出液中的总皂苷质量浓度,以流份份数与流出液中总皂苷质量浓度做图,结果见图1。可见在上样液的质量浓度为0.2 g生药/mL时,在第5个流份开始泄漏,因此确定最大上样量为5BV。
2.3.4 洗脱剂体积分数的确定:精密吸取上样液100 mL,以2 mL/min的流速连续通过HPD300树脂柱,先用4 BV水冲洗,再依次用10%、30%、50%、70%、90%乙醇洗脱,洗脱剂用量均为3 BV,每1 BV接收一次流出液,测定各段流出液中的总皂苷的质量浓度,结果见图2。可见甘草总皂苷主要集中在50%~70%乙醇洗脱部分。
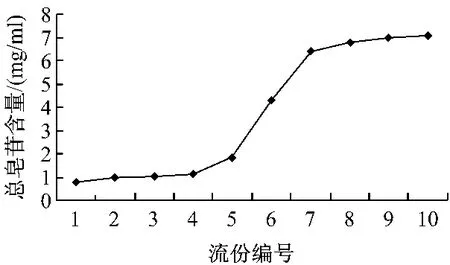
图1 HPD300树脂吸附总皂苷的动态吸附曲线
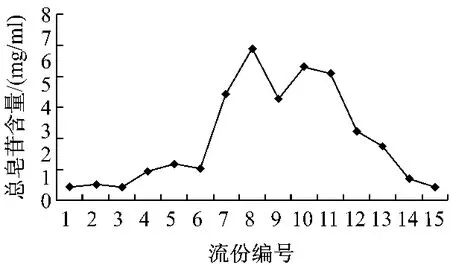
图2 洗脱剂体积分数对动态洗脱性能的影晌
2.3.5 洗脱剂用量的确定:为进一步确定乙醇体积分数并考察洗脱剂用量,取100 mL上样液分别上柱,均用水洗脱4 BV后,分别用50%和70%乙醇洗脱,控制流速2 mL/min,直至流出液无色,每1 BV接收一次流出液,测定各段流出液中的总皂苷的质量浓度,结果见图3。可见50%乙醇洗脱效果好于70%乙醇,且在第6份洗脱液中总皂苷已经很少,可认为树脂柱上吸附的总皂苷已经洗脱完全,故确定洗脱剂为50%乙醇,用量为6 BV。
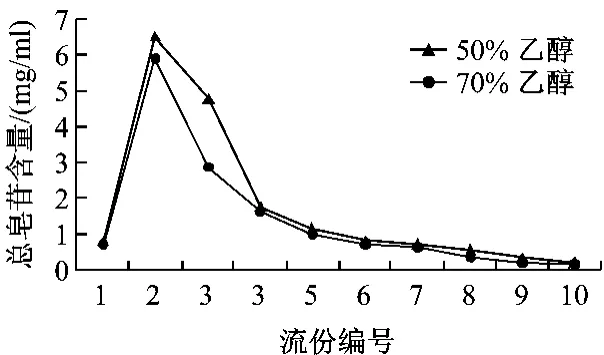
图3 不同体积分数乙醇的洗脱曲线
3 讨论
本实验采用正交试验确定了甘草总皂苷的水煎煮提取条件为加水量8倍,煎煮时间2 h,煎煮次数3次。在这种条件下,甘草总皂苷提取率能达到4.2%。
实验表明HPD300型大孔吸附树脂对甘草总皂苷具有良好的吸附性能,具有较大的吸附量,且易吸附、易解吸。其工艺条件为上样液质量浓度为0.2 g生药/mL,上样量5 BV;50%乙醇洗脱6 BV。
实验中确定的总皂苷提取纯化工艺可为进一步提取纯化甘草总皂苷提供借鉴,从而为进一步开发甘草有效部位新药提供参考。
[1]中国药典[S]. 一部.2005:59-60.
[2]兰 霞,王洪新.比色法测定甘草中总皂苷的含量[J].时珍国医国药,2007,18(4):886-887.
[3]Van Rossum T G,Vulto A G,De Man R A,et al.Review article:glycyrrhizin as a potential treatment for chronic hepatitis[J].Aliment Pharmacol Ther,1998(12):199-205.
[4]王亚红,李战英.甘草酸的提取和精制法研究现状[J].天津化工,2003,17(6):34-36.
[5]郭锦棠,杨俊红,李雄勇,等.微波与索氏提取甘草酸的正交实验研究[J]. 中国药学杂志,2002,37(12):919-922.
[6]王伟明,朱文全,张志波.甘草有效成分提取分离方法的研究进展[J].黑龙江医药,2010,23(1):93-94.
[7]张 嫱,杨振华.微波辅助提取甘草酸的工艺[J].食品研究与开发,2010,31(4):38-40.
[8]张海燕,李 伟,范彩玲,等.超声波法提取甘草中的甘草酸[J]. 河南科学,2009,27(9):1069-1071.
[9]王 颖,孙 晶,张东杰.超临界CO2萃取甘草酸的工艺研究[J].食品与机械,2006,22(3):34-36.
[10]黄明进,王文全,沈寿茂.甘草总黄酮和总皂苷成分的提取工艺及其含量分析[J].中国现代中药,2010,12(4):24-27.
[11]刘玉红,崔红梅,陈 燕,等.正交试验法研究甘草的提取工艺[J].中成药,2004,26(3):239-241.
[12]吴群英,蒋柏泉,白兰莉.大孔树脂对甘草酸的吸附纯化[J]. 江西化工,2006(3):99-101.
[13]袁怀波,叶 明,刘文宏.甘草总三萜酸的大孔树脂分离纯化[J]. 食品工业科技,2008(2):212-214.
猜你喜欢
昆明医科大学学报(2021年6期)2021-07-31 07:39:56
陶瓷学报(2020年6期)2021-01-26 00:38:14
制造技术与机床(2018年10期)2018-10-13 06:36:56
中成药(2018年6期)2018-07-11 03:01:10
中成药(2017年8期)2017-11-22 03:18:44
特产研究(2016年3期)2016-04-12 07:16:17
化工设计通讯(2016年4期)2016-03-13 10:16:21
中国糖料(2014年2期)2014-01-20 07:45:02
经济林研究(2013年3期)2013-04-10 08:27:44
食品科学(2013年6期)2013-03-11 18:20:12
