
稀燃汽车尾气中氮氧化物的催化消除技术
2011-04-09 08:43:37潘广宏
化学工业与工程 2011年3期
潘广宏,孟 明
(天津大学化工学院 天津市应用催化科学与工程重点实验室,天津 300072)
随着现代经济的快速发展,人民生活水平显著提高,汽车保有量也迅速增加。然而汽车在给人们生活带来极大方便的同时,也造成了极大的环境污染。汽车尾气污染已占到全部大气污染的65%~80%。汽车尾气中的主要成份有:碳氢化合物(HC)、氮氧化合物(NOx)、碳氧化物 (COx)和颗粒物(PM)等。这些污染物会引起酸雨、光化学烟雾,温室效应和臭氧空洞效应等许多污染问题,给人类及其赖以生存的环境带来了极大的危害。目前,三效催化剂可以将传统的汽油发动机尾气中的污染物成功地催化消除,但对稀燃发动机尾气中的氮氧化物,三效催化剂效率极为低下。因此,如何高效地消除稀燃汽车尾气中的氮氧化物已迫在眉睫,并已成为环境催化领域的挑战性课题之一。对稀薄燃烧技术进行了简介,并对当前最常用的稀燃NOx催化消除技术,即NO直接分解、选择性催化还原(SCR)和储存还原(NSR)进行了详细评述,同时对未来该领域的发展态势进行了展望。
1 限制汽车尾气污染物排放量的标准
目前,全世界汽车尾气排放标准主要有3大体系[1],即美国、欧洲和日本体系,表1显示了欧盟关于汽车尾气排放标准的具体数据,从表1中可以看出:汽车尾气排放标准越来越严格,总的理念是实现从低排放到超低排放,最终实现零排放。欧盟20世纪90年代开始实行欧洲标准,2000年实施欧III标准,2005年实施欧IV标准,2009实施欧V标准[2]。我国主要采用和借鉴欧洲标准,1999年,我国北京市率先采用欧I排放标准,2004年实施欧II标准,2005年实施欧III标准,2010年与欧盟接轨。
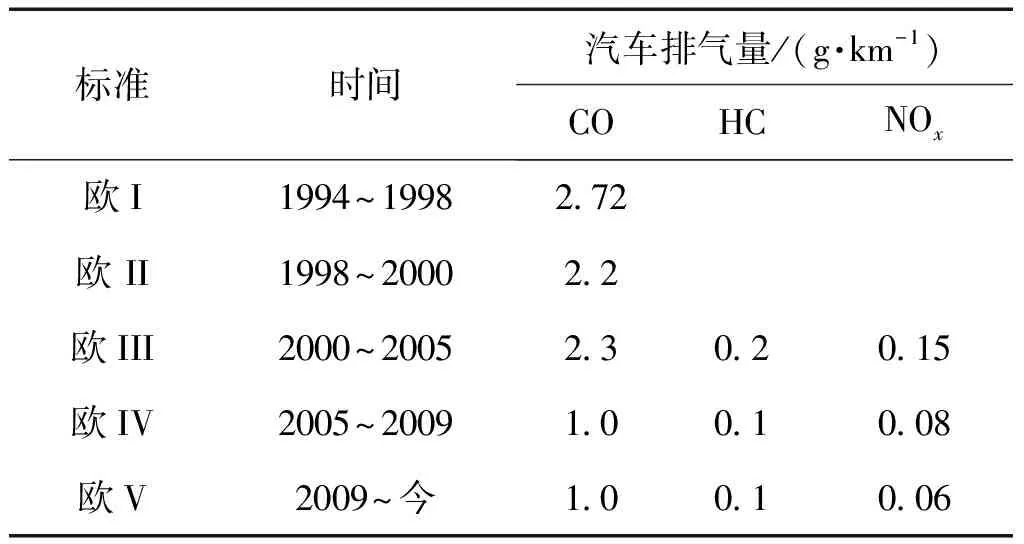
表1 欧洲汽车尾气排放标准Table 1 The European standards of the car emission
2 稀燃技术
消除汽车尾气的方法主要包括机内净化和机外净化。机内净化主要是通过提高燃料的质量和改善发动机的结构,从而改善燃料的燃烧条件,从源头上控制污染物的生成,这是一种比较彻底的净化方式,但是难度很大,其中改善发动机的结构目前进入瓶颈状态;机外净化是汽车尾气的后处理技术,主要指通过安装尾气净化器和尾气消除催化剂将尾气中的CO、HC和NOx等污染物转化为无污染的CO2、H2O及N2。稀燃技术是机内净化的一种,其燃料的燃烧效率高,经济性好,能有效减少温室气体CO2的排放量,降低汽车尾气污染,从节能减排的理念考虑,应该大力发展稀燃机动车[3],许多国家已经开发出了稀燃发动机,并投入汽车市场。
然而在稀燃发动机工作时,空燃比远远偏离了理论空燃比,尾气中O2大量过剩。传统的三效催化剂(three way catalyst简称TWC)只有在空燃比处于理论空燃比(14.6±0.25)附近时,才能够同时使汽车尾气中的CO催化氧化生成CO2,HC催化氧化生成CO2和H2O,NOx催化还原生成N2[4]。在稀燃工况下,传统的三效催化剂只能催化氧化HC和CO生成CO2和H2O,不能有效地消除NOx。因此,如何在稀燃条件下有效地消除NOx成为当前研究的热点,也是整个机外净化技术的新机遇和新挑战。
目前在稀燃技术下脱除NOx的技术主要有NO直接分解、选择性催化还原(SCR)、NOx储存还原(NSR)等。
3 NO直接分解法
NO直接分解法就是通过催化剂的作用将NO直接分解为无毒无害的N2和O2,这一分解反应在热力学上是可行的,但是其反应的活化能高达364 kJ/mol[5],实际上很难实现。因此,这项技术的关键就是寻找合适的催化剂来降低反应的活化能,使反应在较温和的条件下进行。
目前报道的用于NO直接分解反应的催化剂主要有贵金属、金属氧化物、复合氧化物和分子筛型等。贵金属催化剂[6]主要是Pt、Pd和Au等,如Pt/Al2O3,主要的反应活性位是Pt0,NO分解为吸附态的氧原子和氮原子,当温度低于773 K时,吸附态氧原子不容易脱附,随着氧原子覆盖率的增加,催化剂的活性中心被逐渐覆盖,催化剂的活性就逐渐降低[7]。金属氧化物催化剂主要有Co3O4、CuO、Fe2O3和ZrO2等,主要的反应活性中心是金属氧化物表面的氧缺陷[8]。钙钛矿型复合氧化物在高温下结构依然很稳定,但容易脱附氧形成氧空位,形成带负电的表面活性位,但在稀燃条件下,氧气大量过剩时,就会发生“氧抑阻”(oxygen positioning)现象,钙钛矿复合氧化物的表面很容易吸附氧从而使活性降低[9]。Iwamoto等[10]报道分子筛中Cu-ZSM-5的活性最高,NO的转化率能达到90%,但在稀燃条件下,受O2、SO2和H2O的影响严重,很难寻找到具有实际应用价值的NO直接分解催化剂。
4 NOx选择催化还原技术(SCR)
SCR催化剂是在氧气大量过剩时使还原剂优先与NOx反应生成无害的N2的一种方法。SCR催化剂主要有贵金属、金属氧化物、复合氧化物和新型分子筛等催化剂。贵金属型SCR催化剂主要是将Pt、Pd等贵金属负载在SiO2、Al2O3等载体上,此类催化剂具有良好的低温活性,较强的抗硫和抗水中毒能力,但是操作温度范围比较窄[11]。单金属氧化物型SCR催化剂主要有Al2O3、TiO2、ZrO2、SiO2、Cr2O3、Fe2O3和Co3O4等,单金属氧化物型SCR催化剂一般活性不高,需要负载一些活性组分[12]。复合氧化物型SCR催化剂主要有稀土钙钛矿型(ABO3)和铜铁矿结构(ABO2)型,其活性较高,但抗硫能力比较差[13]。
选择性催化还原技术常用的还原剂包括烃类、NH3、尿素、H2和CO等,所用还原剂的类别直接与NOx的脱除效率相关。用烃类化合物为还原剂,可以不用外加还原剂,直接利用汽车尾气中未完全燃烧的烃类物质进行还原,比选用其它还原剂安全系数高,所以稀燃条件下用烃选择催化还原NOx的SCR技术具有很好的开发前景。用NH3作为还原剂[14-15]来消除尾气中NOx,这类催化剂在潮湿、富燃的条件下仍然能具有较高的活性、较长的寿命和较好的抗硫性能,但是外加的还原剂NH3潜在危险系数比较大,可选用尿素作为氨源,尿素通过水解释放出氨,氨与NOx发生氧化还原反应生成无害的N2。选用H2和CO作为还原剂时,NOx的消除率也比较高,富氧条件下以H2和CO共同作为还原剂时,NO的转化率可达到90%以上[16]。
5 NOx储存还原(NSR)技术
5.1 NOx的储存机理
NSR技术是由日本丰田汽车公司首先提出来的在稀燃条件下消除NOx的一种方法[17],稀燃汽油机在稀燃和富燃条件下交替运行,首先经过几十秒的稀燃模式,这个阶段氧气大量过剩,NOx以硝酸盐或亚硝酸盐的形式储存在催化剂的储存组分中,随后快速转换至几秒的富燃模式,这个阶段还原剂过量,在富燃阶段储存的NOx释放出来,并在氧化组分表面被还原成无害的N2,这样循环往复交替进行。NSR催化剂在稀燃阶段NOx的储存路径主要有2种:1种是硝酸盐路径,1种是亚硝酸盐路径。图1显示了NOx的储存路径[18]。硝酸盐路径:主要是尾气中NO在Pt位上被氧化为NO2,接着NO2溢流到Ba位上,与Ba相互作用形成硝酸钡,同时放出NO;亚硝酸盐路线:主要是在尾气中NO在Pt位上被氧化为NO2并直接与相邻的Ba作用形成亚硝酸盐储存下来,然后进一步被氧化成硝酸盐。储存路径与Pt和Ba接触量有关,当相临的Pt和Ba较多时,亚硝酸盐路线就会占主导地位,反之硝酸盐路线就会占主导地位。
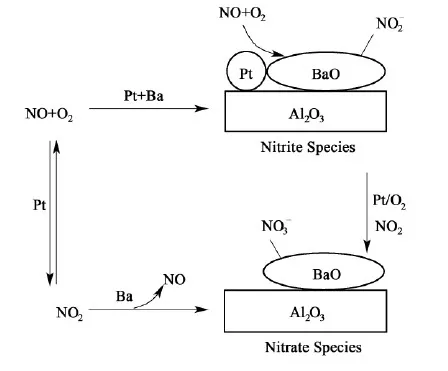
图1 Pt/Ba/Al2O3 催化剂体系中NOx的2种不同储存路径Fig.1 The schematic diagram of the two different NOx storage ways in the Pt/Ba/Al2O3 catalysts
5.2 贵金属型NSR催化剂
Pt/Ba/Al2O3(简称为PBA)是最早开发的NSR催化剂[19],活性组分是Pt,储存组分是碱金属Ba,载体是Al2O3。随着研究的逐步深入,活性组分增加了Pd、Rh和Ag等,储存组分增加了K、Li、Na等多种碱土金属和碱金属,载体增加了SiO2、CeO2、ZrO2以及二元或三元的复合金属氧化物等。贵金属型NSR催化剂的3个组成部分相辅相承,共同决定催化剂的综合性能。
贵金属是NSR催化剂的氧化还原活性中心,在稀燃阶段的储存过程中,它起氧化作用,将NO氧化为NO2,在富燃阶段的还原阶段将脱附的NOx还原为无害的N2。贵金属的前驱体盐的种类、存在状态、粒径分布、负载量、制备方法、与载体的相互作用、与储存中心的距离等都影响其氧化/还原能力。Pt对NO的捕获和氧化能力都很好,被广泛运用为NSR催化剂的活性组分。有报道称Pd在NSR催化剂中比Pt具有更好的活性和热稳定性[20],而且与Pt相比,Pd的价格更便宜,资源也更丰富。从抗硫性能上看,Pt/Rh双贵金属催化剂的抗硫性能最好[21]。
贵金属型NSR催化剂中最常用的储存组分是碱土金属Ba[22],它有很多物种,其最主要的储存物种的储存能力顺序为BaO>Ba(OH)2>BaCO3,这主要是因为各物种的碱性顺序为BaO>Ba(OH)2>BaCO3。
常用的载体[23]有Al2O3、SiO2、CeO2、ZrO2和TiO2等,Al2O3、SiO2、ZrO2等能有效提高催化剂的比表面积和NOx储存能力,但易与活性组分发生反应,热稳定性差,抗硫能力差。TiO2的抗硫能力强,但是比表面积和孔容小。与单一氧化物载体相比复合氧化物(如TiO2-ZrO2)具有更好的储存和抗硫能力,刘咏等[24-25]发现把活性组分Pt和储存组分K负载在不同温度焙烧的载体上后,催化剂的储存和抗硫性能相差很大,当焙烧温度从500 ℃升高到800 ℃时,比表面积显著下降,载体酸量明显减少,表面形成高分散的K2CO3,NOx储存能力逐渐升高,抗硫性能逐渐降低,储存物相主要为自由硝酸根离子,硫化物种主要是形成了体相硫酸盐。邹志强等[26]研究了用TiO2-ZrO2-Al2O3三元复合氧化物作载体的NSR催化剂的储存和抗硫能力,把Al2O3引入到TiO2-ZrO2二元复合氧化物载体中可以大大提高催化剂的比表面积,当n(Al)/n(Ti+Zr)为3时,催化剂的抗硫性能相对最好。
贵金属型NSR催化剂的缺点是:成本高,热稳定性差,易烧结等。
5.3 钙钛矿型NSR催化剂
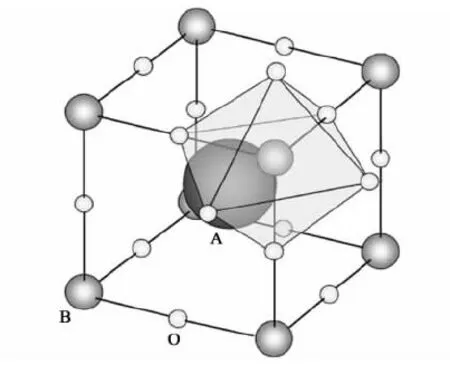
图2 钙钛矿复合氧化物(ABO3)的晶体结构Fig.2 The crystal structure of the perovskite (ABO3)
钙钛矿型复合氧化物(ABO3)的结构与矿石CaTiO3的晶体结构类似,其结构如图2[27]所示。晶体中A位金属离子与氧形成12配位,一般半径比较大,位于八面体所构成的空穴中,通常为碱金属、碱土金属以及镧系金属,B位金属离子半径相对小些,被氧所包围,通常为3d、4d和5d的过渡金属离子,氧分布在八面体的顶点处。
钙钛矿ABO3的结构可以调变,可以形成有缺陷的钙钛矿,可以通过A位或B位掺杂或取代来产生A位或B位阳离子空位或变价,或形成氧空位等[28]。
钙钛矿结构稳定,具有良好的氧化还原能力,结构可调变,晶格中可以固定一定尺寸范围内的粒子,具有良好的热稳定性等,因此钙钛矿是很有应用前景的NSR催化剂材料。
Wang H等[29]研究了LaBO3(B=V,Cr, Mn, Fe, Co, Ni, Cu)对NOx和soot同时消除的能力,研究发现氧化能力依次为LaNiO3≈ LaCoO3>LaMnO3>LaFeO3>LaCro(3);Arai H等[30]报道了含钡的BaBO3(B=Mn, Fe, Co, Ni)钙钛矿通过吸附/脱附循环消除NOx。Milt等[31-32]研究了焙烧温度对BaCoO3钙钛矿结构和储存性能的影响,发现随着焙烧温度的升高,有利于形成缺陷钙钛矿,在400 ℃焙烧时还没有形成完整的钙钛矿晶形,主要物相为Ba(NO3)2和Co3O4,当焙烧温度升高到700 ℃时,可以形成化学计量的完整BaCoO3钙钛矿,有一定的储存能力;当焙烧温度进一步升高到1 000 ℃后,形成缺陷钙钛矿BaCoO2.74,有良好的储存能力。李新刚等[33]也研究了BaFeO3钙钛矿的NOx储存性能,发现Ba-Fe-O拥有良好的抗硫能力,350~400 ℃为NOx的最优储存温区。Kaddouri等[34]研究了 La-Ce-Mn-O钙钛矿,发现La-Ce-Mn-O拥有良好的氧化和抗硫性能。
Hodjati等[35-36]发现:当A位为Ba离子不变,B位离子分别为Sn、Zr和Ti时,NOx储存能力为Sn>Zr>Ti,主要原因是BaSnO3中B—O键能最弱;当B位为Zr离子不变,A位离子分别为碱土金属中的Ca、Sr和Ba时,NOx储存能力为Ba>Sr>Ca,主要原因是BaSnO3中A位元素的正电性最高。吸附NOx的主要形态是离子硝酸盐和N键合硝酸盐(O—Ba—NO2),钙钛矿中存在自由氧空位与Lewis酸NO2发生反应形成N键合硝酸盐O—Ba—NO2,温度影响二氧化氮吸附物种离子硝酸盐和N键硝酸盐的比例。NOx在钙钛矿结构上吸/脱附过程的机理为[37]:部分钙钛矿结构在吸附过程中打开与NO2形成硝酸盐和N键合硝酸盐;在脱附过程中再重新形成钙钛矿结构。在反应气中加入25×10-6的SO2,经过15个贫燃-富燃循环,BaSnO3催化剂完全失活,体相硫酸盐峰的强度随温度的升高而增强[38]。可见,温度越高,BaSnO3钙钛矿越容易硫中毒。
钙钛矿的主要缺点是比表面积较小,比表面积一般在10 m2/g以下,因此如何制得大比表面积的钙钛矿是研究的热点,目前提高催化剂的比表面积的主要途径有2种:1)通过柠檬酸络合溶胶凝胶,冷冻干燥法,化学气相沉积法,微乳液法,水热合成法等技术制备出大比表面积的钙钛矿[39];2)将钙钛矿负载在大比表面积的载体上。但是高温焙烧时,钙钛矿的前驱体容易与载体发生相互作用,形成杂相,阻碍钙钛矿的形成。因此制备负载型钙钛矿时载体的选择至关重要,目前常用的载体[40]主要包括金属氧化物、堇青石、复合氧化物、水滑石、蜂窝载体和介孔分子筛等。当选用大比表面积的Al2O3做载体时,可先用涂层技术在载体的表面涂一层惰性物质(如La2O3),否则很难制备成功[41];块状堇青石的结构有利于钙钛矿的附着;TiO2-ZrO2在高温下可以形成具有较大比表面积的结构稳定的固溶体,是一种被看好的载体;SBA-15或者ZSM-5分子筛结构稳定,有介孔结构,比表面积大,是有应用前景的载体。
6 问题与展望
稀燃技术优势突出,随着科技的进步,稀燃技术必然会被广泛应用。NOx储存还原将成为治理稀燃NOx污染的核心技术。但现有的NSR催化剂还有诸多不足,如传统的Pt/Ba/Al2O3NSR催化剂需要不断地提高其抗硫性能和抗水性能;贵金属型NSR催化剂活性好,但是其成本高,热稳定性也有待进一步提高;钙钛矿是一种很好的代替贵金属的材料,其成本低,活性好,但是抗硫性能差,而且在还原气氛下,钙钛矿结构容易被破坏。未来需要围绕以下几个方面开展研究: 1)新型NOx储存剂的研发; 2)具有较大比表面积和较高热稳定性的复合酸性载体的制备;3)低贵金属NSR催化剂的研发,寻找可以部分取代或全取代贵金属的替代材料,以降低催化剂的成本。这些方面的研究工作将使NOx储存还原技术具有更加光明的应用前景。
参考文献:
[1]SOUNAK R, BAIKER A. NOxstorage-reduction catalysis: From mechanism and materials properties to storage-reduction performance[J]. Chem Rev, 2009, 109(9): 905-915
[2]ANNEMANS L, STRENS D, LOX E,etal. Cost-effectiveness analysis of aprepitant in the prevention of the motherapy-induced nausea and vomiting in Belgium[J].Supportive Care in Cancer, 2008, 16(8): 905-915
[3]CIAMBELLI P, CORBO P, GAMBINO M. Catalysis in environmental applications selected papers from the international symposium on catalysis and zeolites[J]. Catal Today, 1995, 26 (1): 33-39
[4]LUO J, MENG M, ZHA Y. A comparative study of Pt/Ba/Al2O3and Pt/Fe-Ba/Al2O3NSR catalysts: New insights into the interaction of Pt-Ba and the function of Fe[J]. Appl Catal B, 2008, 78(1/2): 38-52
[5]SAVIKHIN S, ZHU Y, BLANKENSHIP R E,etal. Ultrafast energy transfer in chlorosomes from the green photosynthetic bacterium chloroflexus aurantiacus[J]. J Phys Chem, 1996, 100 (9): 3 320-3 322
[6]BELANGER R, MOFFAT J B. Removal of NO2from gaseous streams by sorption and conversion on 12-tungstophosphoric acid[J]. Environ Sci Technol, 1995, 29 (6): 1 681-1 685
[7]AMIRNAZMI A, BOUDART M. Decomposition of nitric oxide on platinum[J]. J Catal, 1975, 39 (3): 383-394
[8]WINTER E R S. The catalytic decomposition of nitric oxide by metallic oxides[J]. J Catal, 1971, 22 (2): 158-170
[9]TOFAN C, KLVANA D, KIRCHNEROVA J. Decomposition of nitric oxide over perovskite oxide catalysts: Effect of CO2, H2O and CH4[J]. Appl Catal B, 2002, 36 (4): 311-323
[10]CHOI B C, FOSTER D E. State of the art of de-NOxtechnology using zeolite catalysts in automobile engines[J]. J ind eng chem, 2005, 1: 1-9
[11]QI G, YANG R, THOMPSON L. Catalytic reduction of nitric oxide with hydrogen and carbon monoxide in the presence of excess oxygen by Pd supported on pillared clays[J]. Appl Catal A, 2004, 259 (2): 261-267
[12]FERRI D, FORNI L, DEKKERS M A P H,etal. NO reduction by H2over perovskite-like mixed oxides[J]. Appl Catal B, 1998, 16 (4): 339-345
[13]CENTI G, PERATHONER S. Nature of active species in copper-based catalysts and their chemistry of transformation of nitrogen oxides[J]. Appl Catal A, 1995, 132(2): 179-259
[14]CHOUNG J W, NAM I S, HAM S W. Effect of promoters including tungsten and barium on the thermal stability of V2O5/sulfated TiO2catalyst for NO reduction by NH3[J]. Catalysis Today, 2006, 111(3/4): 242-247
[15]SULLIVAN D H, HAROLD M P, CONNER JR W C. Catalyst transformation during NO+NH3reaction over vanadium oxide using in situ FTIR emission spectroscopy[J]. J Catal, 1998, 178(1): 108-118
[16]PITCHON V, FRITZ A. The relation between surface state and reactivity in the de-NOxmechanism on platinum-based catalysts[J]. J Catal, 1999, 186 (1): 64-74
[17]LEI C, QING F. Effect of BaO morphology on NOxabatement: NO2interaction with unsupported andγ-Al2O3-supported BaO[J]. J Phys Chem C, 2008, 112(43): 16 924-16 931
[18]FORZATTI P, CASTOLDI L, NOVA I. NOxremoval catalysis under lean conditions[J]. Catal Today, 2006, 117 (1/3): 316-320
[19]NOVA I, LIETTI L, CASTOLDI L,etal. New insights in the NOxreduction mechanism with H2over Pt-Ba/γ-Al2O3lean NOxtrap catalysts under near-isothermal conditions[J]. J Catal, 2006, 239 (1): 244-254
[20]ROY S, MARIMUTHU A, HEGDE MS,etal. Pd ion substituted CeO2: A superior de-NOxcatalyst to Pt or Rh doped ceria[J]. Catal Commun, 2008, 26 (9): 811-815
[21]AMBERNTSSON A, FRIDELL E, SKOGLUNDH M. Influence of platinum and rhodium composition on the NOxstorage and sulphur tolerance of a barium based NOxstorage catalyst[J]. Appl Catal B, 2003, 46 (3): 429-439
[22]KIM DH, J SZANYI, JH KWAK,etal. Effects of sulfation level on the desulfation behavior of pre-sulfated Pt BaO/Al2O3lean NOxtrap catalysts[J]. J Catal, 2009, 113 (17): 7 336-7 341
[23]VERRIER C L M, KWAK J H, KIM D H,etal. NOxuptake on alkaline earth oxides (BaO, MgO, CaO and SrO) supported on Al2O3[J]. Catalysis Today, 2008, 136 (1/2): 121-127
[24]刘咏,孟明,郭丽红,等. 载体焙烧温度对NOx储存还原催化剂K/Pt/TiO2-ZrO2结构与性能的影响[J]. 催化学报,2007,28 (10): 850-856
[25]LIU Y, MENG M, LI X G,etal. NOxstorage behavior and sulfur-resisting performance of the third-generation NSR catalysts Pt/K/TiO2-ZrO2[J]. Chem Eng Res Des, 2008, 86: 932-940
[26]ZOU Z Q, MENG M, ZHOU X Y,etal. The effect of Al2O3doping into TiO2-ZrO2on the storage and sulfur-resistance performance of the NOxtrap catalyst Pt/K/TiO2-ZrO2[J]. Catal Lett, 2009, 128(3/4): 475-482
[28]TANAKA H, MISONO M. Advance in designing perovskite catalysts[J]. Current Opinion in Solid State and Materials Science, 2001, 5(5): 381-387
[29]WANG H, ZHAO Z, XU CM,etal. Simultaneous removal of soot particles and NO from diesel engines over LaBO3perovskite-type oxides[J]. J Phys Chem, 2008, 29(7): 649-654
[30]ARAI H, MACHIDA M. Removal of NOxthrough sorption-desorption cycles over metal oxides and zeolites[J]. Catal Today, 1994, 22(4): 97-109
[31]MILT V G, ULLA M A, MIRO E E. Abatement of diesel exhaust pollutants: NOxadsorption on Co,Ba,K/CeO2catalysts[J]. J Catal, 2003, 220(2): 424-432
[32]MILT V G, ULLA M A, MIRO E E. NOxtrapping and soot combustion on BaCoO3perovskite: LRS and FTIR characterization[J]. Appl Catal B, 2005, 57(6): 13-21
[33]KADDOURI A, GELIN P, DUPONT N. Methane catalytic combustion over La-Ce-Mn-O perovskite prepared using dielectric heating[J]. Catal Commun, 2009, 10: 1 085-1 089
[34]陈加福,孟明,林培琰,等. BaFeO3和BaCeO3钙钛矿型氧化物的储氮性能[J]. 催化学报, 2003, 24 (6): 419-422
[35]HODJATI S, VAEZZADEH K, PETIT C. Absorption/desorption of NOxprocess on perovskites: performances to remove NOxfrom a lean exhaust gas[J]. Appl Catal B, 2000, 26 (1): 5-16
[36]HODJATI S, PETIT C, PITCHON V. Absorption/desorption of NOxprocess on perovskites: impact of SO2on the storage capacity of BaSnO3and strategy to develop thioresistance[J]. Appl Catal B, 2001, 30 (3/4): 247-257
[37]COURSON C, KHALFI A, MAHZOUL H,etal. Experimental study of the SO2removal over a NOxtrap catalyst[J]. Catal Commun, 2002, 3 (10): 471-477
[38]HODJATI S, PETIT C, PITCHON V. Absorption/desorption of NOxprocess on perovskites nature and stability of the species formed on BaSnO3[J]. Appl Catal B, 2000, 27 (2): 117-126
[39]SHU J, KALIAGUINE S. Well-dispersed perovskite-type oxidation catalysts[J]. Appl Catal B, 1998, 16 (4): 303-308
[40]BIALOBOK B, TRAWCZYNSKI J, MISTA W. Ethanol combustion over strontium and cerium-doped LaCoO3catalysts[J]. Appl Catal B, 2007, 72 (4): 395-403
[41]NITIN K, LABHSETWAR A, WATANABE R. Alumina supported, perovskite oxide based catalytic materials and their auto-exhaust application[J]. Appl Catal B, 2001, 33(1): 165-173
猜你喜欢
证券市场周刊(2024年13期)2024-04-16 04:33:35
贵金属(2021年1期)2021-07-26 00:39:20
贵金属(2021年1期)2021-07-26 00:39:20
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26 00:58:18
中国资源综合利用(2017年2期)2018-01-22 02:45:06
物理学进展(2017年1期)2017-02-23 01:35:44
云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:14
环境科技(2015年5期)2015-11-08 12:09:10
环境科技(2015年5期)2015-11-08 12:09:08
太阳能(2015年4期)2015-02-28 17:08:19
