
固相萃取-超高效液相色谱-质谱/质谱法测定动物源性食品中二硝甲酚残留量
2011-04-06 09:40:49康庆贺杨长志高凯洋
食品科学 2011年14期
吴 岩,康庆贺,刘 永,胡 珅,杨长志,高凯洋
(1.黑龙江出入境检验检疫局,黑龙江 哈尔滨 150001;2.黑龙江大学化学化工与材料学院,黑龙江 哈尔滨 150086;3.东宁出入境检验检疫局,黑龙江 东宁 157200)
固相萃取-超高效液相色谱-质谱/质谱法测定动物源性食品中二硝甲酚残留量
吴 岩1,2,康庆贺1,*,刘 永1,胡 珅3,杨长志1,高凯洋2
(1.黑龙江出入境检验检疫局,黑龙江 哈尔滨 150001;2.黑龙江大学化学化工与材料学院,黑龙江 哈尔滨 150086;3.东宁出入境检验检疫局,黑龙江 东宁 157200)
建立动物源性食品中二硝甲酚残留量的分析方法。样品以丙酮-正己烷(1∶2,V/V)为提取溶剂,经高速匀浆方法提取,提取液经Oasis HLB固相萃取柱净化,除去样品中大部分的脂肪和无机盐等干扰基质,有效地降低了样品中的复杂基质所带来的背景干扰。以乙腈-水(梯度洗脱)为流动相经BEH C18柱(50mm×2.1mm,1.7μm)进行分离后以MS/MS多反应监测扫描模式下检测。方法的相关系数r>0.999,最低检出限为0.005mg/kg。在3种添加水平(0.005、0.010、0.020mg/kg)条件下,其平均回收率为82.6%~108%,相对标准偏差小于10%(n=6)。该方法是一种快速、准确、灵敏度高的检测动物源性食品中二硝甲酚残留量的检测方法。
固相萃取;超高效液相色谱-质谱/质谱;二硝甲酚;动物源性食品;除草剂
二硝甲酚,化学名称为4,6-二硝基邻甲酚(dinitro-O-cresol,DNOC)是属于除草剂,常用在春天或冬天耕作时期防除阔叶杂草,同时,由于其具有胃毒和触杀作用,也可做杀虫剂使用[1]。这种农药药效高、易分解,在农药品种中占有极为重要的位置。医学研究表明,二硝甲酚对小鼠的半数致死量LD50为21mg/kg,对人、畜的急性毒性很强,其广泛应用可能对人类造成潜在的健康威胁和生态环境的污染。
目前,美国、欧盟和日本等许多发达国家对二硝甲酚的残留提出严格的限量要求,尤其是日本推行的肯定列表制度,规定其最大残留限量为0.01mg/kg,而目前我国分析动物源性食品中二硝甲酚残留量的方法不多,已有的行业标准检测方法采用将样品中的二硝甲酚与重氮甲烷进行衍生化后经气相色谱-氮磷检测器(GCNPD)测定[2-3],该方法存在用时长、消耗试剂较多、重氮甲烷制备过程繁琐、衍生化条件需控制严格、衍生化效率不稳定、假阳性结果不能准确判断、检测低限(0.02mg/kg)不能满足实际需要需要等实际问题。因此,本实验建立了一种以Oasis HLB固相萃取柱进行净化,采用超高效液相色谱-质谱/质谱法(UPLC-MS/MS)测定动物源性食品中二硝甲酚残留的检测方法,该方法具有操作方便、分析速度快、灵敏度高、选择性强、定性准确等优点,对于弥补该农药检测方法的不足、解决国外贸易技术壁垒、推动食品安全又快又好的发展具有一定的参考意义。
1 材料与方法
1.1 材料与试剂
取猪肉、鸡肉、牛肉及其脏器代表性样品约500g,将其可食部分切碎后,用组织捣碎机将充分捣碎均匀,装入洁净容器作为试样,密封并标明标记,于-18℃以下保存。
丙酮、正己烷、甲醇和乙腈均为色谱纯试剂;无水硫酸钠(分析纯,650℃灼烧4h,贮于密封干燥器中备用);氯化钠(分析纯,200℃烘干2h)。二硝甲酚标准品(纯度大于98%) 德国DR公司。
ENVI Alumina-N固相萃取柱[1000mg(6mL)]、ENVI Florisil固相萃取柱[1000mg(6mL)] 美国Supelco公司;Oasis HLB固相萃取柱[60mg(3mL)] 美国Waters公司。
1.2 仪器与设备
超高效液相色谱-质谱/质谱仪 美国Waters公司;高速匀浆机、旋转蒸发器 德国IKA公司;氮吹仪;高速离心机;固相萃取装置 美国Supelco公司。
1.3 液相色谱-质谱条件
色谱柱:BEH C18(50mm×2.1mm,1.7μm);流动相A为乙腈,B为水;梯度洗脱:0~0.5min,15% A;0.5~1.0min,15%~60% A;1.0~2.0min,60% A;2.0~2.5min,60%~15% A;2.5~3.0min,15% A。流速0.25mL/min;柱温30℃;进样量5μL。
电喷雾离子源:负离子模式;毛细管电压:3.0KV;锥孔电压:40V;离子源温度:110℃;脱溶剂器温度:350℃;脱溶剂气流量:500L/h;锥孔气流量:50L/h。扫描方式:多反应离子监测模式。
1.4 标准溶液的配制
准确称取适量的二硝甲酚农药标准品,用乙腈配制成100.0mg/L的标准储备液;根据需要再用水逐级稀释成质量浓度为0.005、0.01、0.05、0.2mg/L和0.5mg/L的系列混合标准工作溶液。
1.5 样品前处理
称取5g绞碎均匀的样品于50mL的聚丙烯离心管中,加入10mL水、4g氯化钠,再加入20mL丙酮-正己烷(1∶2,V/V),用匀浆机高速均质2min,然后在3500r/min离心5min;将上清液通过无水硫酸钠柱脱水于100mL浓缩瓶中。再用20mL正己烷-丙酮重复提取一次,合并提取液于同一100mL浓缩瓶中,在35℃水浴中用旋转蒸发器浓缩至近干,再用氮气流吹干。向浓缩瓶中加入3mL 20%甲醇溶液溶解残渣待净化。
将Oasis HLB固相萃取小柱依次用3mL甲醇、3mL水进行预淋洗,然后将提取液上样,上样后依次用3mL水、3mL 20%甲醇淋洗净化柱,控制流速在1~2mL/min,弃去淋洗液,然后用2mL甲醇洗脱目标化合物,收集洗脱液与15mL的刻度试管中,在40℃条件下氮气吹至近干,用流动相定容至5mL,待测。
2 结果与分析
2.1 色谱条件的优化
为了优化二硝甲酚的分离条件,本方法分别对BEH C18(50mm×2.1mm,1.7μm)柱与BEH Phenyl(苯基) (50mm×2.1mm,1.7μm)柱的分离效果进行了比较(图1),可以看出,在使用相同流动相时,C18柱的分离效果要远远好于Phenyl柱,由于二硝甲酚的极性较强,反相保留原理的C18柱更适于分离目标化合物。
对于流动相种类的优化,本方法对以乙腈-水体系和甲醇-水进行了比较,两者在分离度灵敏度方面的响应程度差别不大,考虑到乙腈的溶解性较好,本方法还是选用乙腈-水体系作为流动相对二硝甲酚进行分离测定。
从图1可以看出,二硝甲酚的保留时间为1.67min,在保证较高柱效、较高分离度的前提下,采用本方法较已有的气相色谱法在很大程度上提高了分析的速度与效率,节约了分析时间与分析成本。
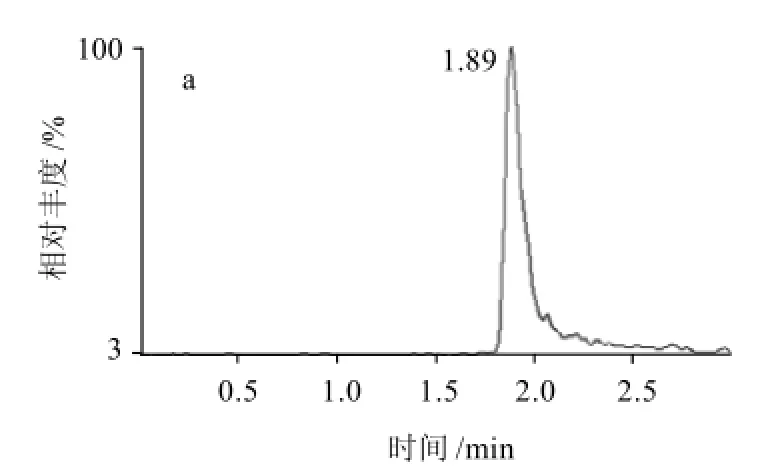

图1 不同色谱柱分离二硝甲酚的色谱图Fig.1 Separation profile of DONC on different chromatographic columns
2.2 提取溶剂的选择
由于二硝甲酚属于属于极性较强的化合物,在选取提取溶剂时也应遵循“相似相容”原理,本方法对乙腈、丙酮、乙酸乙酯、丙酮-正己烷(1∶2,V/V)等提取溶剂的提取效率进行了比较[4]。通过实验可知,在采用乙腈、丙酮作为提取溶剂时,均质的过程中能够和容易的将基质均质完全,但是,提取液中含有一定量的水,在浓缩的过程中即使在较低温度(不高于35℃)时也会出现暴沸现象,并且随着溶剂的蒸发减少,浓缩瓶内出现大量的表面张力较大的泡沫,随时可能倒吸进入蒸发腔内,不仅污染浓缩系统,而且造成目标化合物的损失,蒸发效率不高,无法进行下一步的净化;当采用乙酸乙酯、丙酮-正己烷(1∶2,V/V)作为提取溶剂时,提取液在浓缩时较为容易,但是就提取效率而言,由于乙酸乙酯极性相对丙酮-正己烷(1∶2,V/V)较弱,后者的提取效率更高,作为二硝甲酚的提取溶剂效果更好。
2.3 固相萃取条件的优化
由于动物源性样品基质(动物肉及动物肾脏等)中的脂肪和提取液中的无机盐类是产生的基质效应的主要因素,因此本方法选取能够除去这两种干扰物质的ENVI Alumina-N、ENVI Florisil和Oasis HLB固相萃取柱的净化效果进行优化比较[5]。
采用ENVI Alumina-N和ENVI Florisil净化时,二者属正相固相萃取柱,在使用前依次用3mL丙酮、5mL正己烷预活化,提取液经浓缩后用正己烷定容至3mL上样,由于二硝甲酚的化学结构上带有两个硝基基团,具有较强的电负性,理论上能够在非极性溶剂(使用正己烷作为上样溶剂)上样时被固定相吸附,在极性溶剂条件下[采用丙酮-正己烷(1∶9,V/V)作为洗脱溶剂]能够被解吸,但是实验表明,在作淋洗曲线时,上样和洗脱的过程中都有二硝甲酚流出,并且,当洗脱液的体积达到20mL时还没有将其完全洗脱下来,不符合正相色谱的吸附解吸理论,得不到很好的净化脂肪的效果。
当采用Oasis HLB固相萃取柱时,依次采用3mL甲醇、3mL水进行活化后,将待测溶液以20%甲醇溶液上样,二硝甲酚能够完全被吸附保留在固定相上,然后用3mL水淋洗该柱,除去基质中的水溶性化合物和无机盐类等干扰物质,再用3mL 20%的甲醇溶液淋洗除去极性较强的小分子有机酸类干扰物质,最后用2mL甲醇洗脱目标化合物,通过以上淋洗步骤,能够较好地降低样品基质效应所带来的干扰,得到满意的净化效果(图2),对提高仪器的灵敏度有明显的作用。
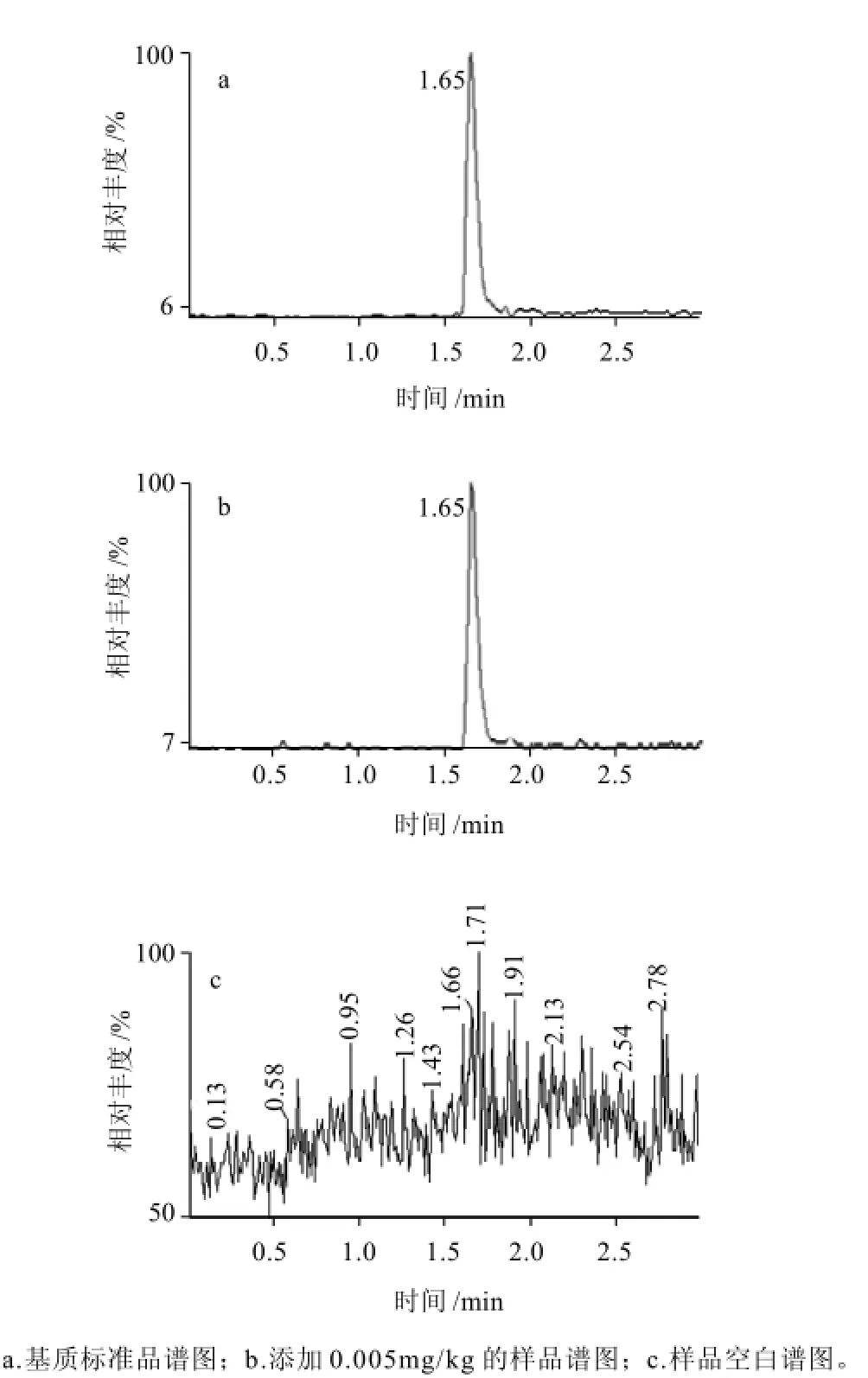
图2 猪肉样品经Oasis HLB固相萃取柱净化后的色谱图Fig.2 Chromatogram of pork sample cleaned up on Oasis HLB solid-phase extraction column
2.4 MS/MS特征离子对的选择
由于二硝甲酚在苯环结构上的4位和6位上含有电负性的硝基基团,故在优化质谱条件时选择负源进行电离[6],本方法采用乙腈-水为流动相,结合基质空白和基质标液的离子扫描图进行了优化,通过优化毛细管电压与碰撞能量,找到以M-H为母离子的4组特征离子对分别为m/z 196.68>179.74、196.68>166.69、196.68>136.81、196.68>108.53,其裂解碎片图见图3。
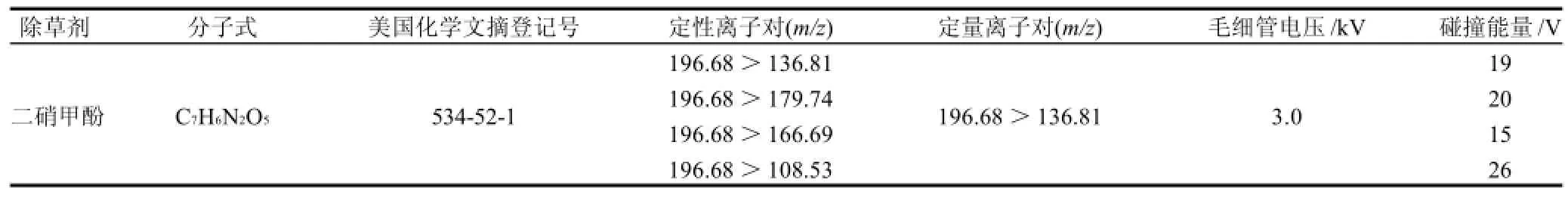
表1 二硝甲酚的质谱分析优化参数Table 1 Optimal parameters for the MS/MS analysis of DNOC residue in the MRM mode

图3 二硝甲酚的裂解原理图Fig.3 Fragmentation principle of DNOC
由裂解碎片的结构可以看出,m/z 196.68>136.81与m/z 196.68>108.53这两组特征离子对的结构稳定,从而确定了二硝甲酚在多反应监测模式下信号的特征离子对的最佳条件参数(表1)。
2.5 标准曲线和最低检出限
配制二硝甲酚的质量浓度分别为0.005、0.01、0.02、0.2、0.5mg/L的标准溶液,进行然后进行UPLC-MS/MS分析。测定时,根据其标准物质和待测样品的MRM离子流图中的峰面积,外标法定量。以农药的选择离子的峰面积(A)对农药的质量浓度C/(mg/L)绘制标准曲线,并求得直线回归方程A=2305.2C+20.1,在0.005~0.5mg/L内的线性关系良好,且相关系数r>0.999,方法的最低检出限为0.005mg/kg,能够满足残留检测的要求。
2.6 方法的回收率及精密度
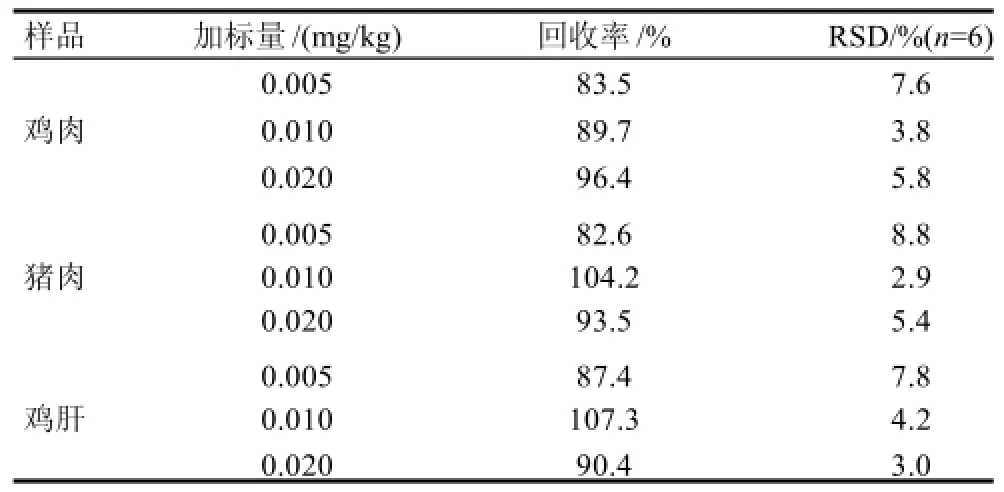
表2 动物源性食品(鸡肉、猪肉、鸡肝)中二硝甲酚添加回收实验结果Table 2 Recovery rates of DNOC in spiked chicken meat, pork and chicken liver
本方法采用标准加入法,选取已粉碎均匀鸡肉、猪肉、鸡肝作为代表性样品分别进行添加回收实验[7-21]。准确称取5.00g样品分别按照3个添加水平(0.005、0.01、0.02mg/kg)添加二硝甲酚标准品,每个浓度平行测定6次,按上述方法提取净化样品,并进行UPLC-MS/MS分析,计算平均回收率和精密度,结果得到所有农药的回收率均在82.6%~108%之间,精密度也均小于10%,具体实验结果见表2。
3 结 论
本研究建立的UPLC-MS/MS方法,样品前处理提取效率高、固相萃取的净化效果较好、条件易于控制,并且采用超高效液相色谱-质谱/质谱分析,分析速度快、选择性好、灵敏度高,极大的缩短了检测时间,结果准确可靠。本方法的回收率、精密度和检出限优于文献报道值和原有的行业标准。本方法的定量限低于日本肯定列表所规定的最高残留限量,可以用于动物源性食品中二硝甲酚残留量的检测。
[1]张禾茂, 黄建炎. 4,6-二硝基邻甲酚合成工艺研究[J]. 安徽化工, 2004, 130(4)∶ 10-11.
[2]杨长志, 陈丽, 康庆贺, 等. 气相色谱法测定鸡肉中二硝甲酚的残留量[J]. 化学工程师, 2003, 94(1)∶ 24-25.
[3]SN 0707—1997 出口肉及肉制品中二硝甲酚残留量检验方法[S].
[4]张莹, 黄志强, 李拥军, 等. 气相色谱法测定茶叶中多种拟除虫菊酯类农药残留量[J]. 分析化学, 2002, 30(3)∶ 377.
[5]李樱, 储晓刚, 仲维科, 等. 凝胶渗透色谱和固相萃取净化-气相色谱分离组合法测定糙米中的残留农药[J]. 分析化学, 2004, 32(10)∶1235-1328.
[6]徐锦忠, 张晓燕, 吴斌, 等. 高效液相色谱-串联质谱法同时检测鸡肉和鸡蛋中合成类固醇类激素和糖皮质激素[J]. 分析化学, 2009, 37 (2)∶ 341-346.
[7]刘琪, 孙雷, 张骊. 超高效液相色谱-串联质谱法测定猪肝中有机磷农药残留量的研究[J]. 分析测试学报, 2010, 29(7)∶ 747-750.
[8]黄捷, 白桂昌, 吕轶峰. 快速高分离液相色谱-串联质谱法测定水产品中敌百虫残留[J]. 中国卫生检验杂志, 2010, 20(7)∶ 1666-1669.
[9]郑军红, 庞国芳, 范春林, 等. 液相色谱-串联质谱法测定牛奶中128种药物残留[J]. 色谱, 2009, 27(3)∶ 254-263.
[10]张乔. 农药污染物残留分析方法汇编[M]. 北京∶ 化学工业出版社, 1990∶ 131-133.
[11]POUCKE C V, PETEGH EM C V. Development and validation of amultianalyte method for the detection of anabolic steroids in bovineurine with liquid chromatography- tandem mass spectrom etry[J]. J Chromatogr∶B, 2002, 772(2)∶ 211-217.
[12]CAI J, HENION J. Quantitative multi residue determination of agonists in bovine urine using on-line immunoaffinity extraction coupled columnpacked capillary liquid chromatography- tandem mass spectrom etry[J]. J Chromatogr∶ B, 1997, 691(2)∶ 357-370.
[13]冯冰, 张佳宜, 王英, 等. 猪肝中莱克多巴胺残留的分析研究[J]. 食品研究与开发, 2007, 28(4)∶ 147-149.
[14]申屠芬琴, 强致懿, 张素霞, 等. 猪肾脏等组织中盐酸菜克多巴胺的HPLC 检测方法及其残留消除[J]. 畜牧兽医学报, 2007, 38(6)∶ 589-590.
[15]臧勇军, 张春晖, 张慧, 等. 液相色谱- 电喷雾质谱同时测定肉类中的莱克多巴胺与克伦特罗[J]. 肉类工业, 2007(1)∶ 38-39.
[16]MATUSZEWSKI B K, CONSTANZER M L, CHAVEZ-ENG C M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS[J]. Analytical Chemistry, 2003, 75 (13)∶ 3019-3030.
[17]MEI H, HSIEH Y, NARDO C, et al. Investigation of matrix effects in bioanalytical high-performance liquid chromatography/tandem mass spectrometric assays∶ application to drug Discovery[J]. Rapid Commun Mass Spectrom, 2003, 17(1)∶ 97-103.
[18]ZROSTLIKOVA J, HAJSLOVA J, POUSTKA J, et al. Alternative calibration approaches to compensate the effect of co-extracted matrix components in liquid chromatography-electrospray ionisation tandem mass spectrometry analysis of pesticide residues in plant materials[J]. J Chromatogr A, 2002, 973(1/2)∶ 13-26.
[19]ANDERSON D B, VEEHUISEN E L, WAITT W P, et al. Effect of ractopamine on nitrogen retention, growth performance and carcass composition of finisher pigs[J]. J Anim Sci, 1987, 65(Suppl 1)∶ 130-131.
[20]WATKINS L E, JONES D J, MOWREY D J, et al. The effect of various levels of ractopamine hydrochloride on the performance and carcass characteristics of finishing swine[J]. J Anim Sci,1990, 68(11)∶3588-3595.
[21]ENGESETH N J, LEE K O, BERGEN W G, et al. Fatty acid profiles of lipid depots and cholesterol concentration in muscle tissue of finishing pigs fed ractopamine[J]. J Food Sci, 1992, 57(5)∶ 1060-1062.
Determination of 4,6-Dinitro-O-Cresol (DNOC) Residue in Animal-origin Foods by Solid-phase Extraction and Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry
WU Yan1,2,KANG Qing-he1,*,LIU Yong1,HU Shen3,YANG Chang-zhi1,GAO Kai-yang2
(1. Heilongjiang Entry-Exit Inspection and Quaratine Bureau, Harbin 150001, China;2. School of Chemistry and Materials Science, Heilongjiang University, Harbin 150086, China;3. Dongning Entry-Exit Inspection and Quaratine Bureau, Dongning 157200, China)
A solid-phase extraction and ultrahigh performance liquid chromatography-tandem mass spectrometry method has been developed for the determination of DNOC residue in animal-origin foods. Samples were extracted with acetone-hexane (1∶2, V/V) after high-speed homogenization. The crude extract was purified on Oasis HLB solid phase extraction column to remove fat and inorganic salts, which could effectively reduce the background interference generated by complex matrix in samples. The DNOC residue was separated on BEH C18 (50 mm × 2.1 mm, 1.7μm) column using acetonitrile-water as the mobile phase by gradient elution and detected using a tandem mass analyzer in the multi-reaction monitoring (MRM) mode. The developed method exhibited an excellent linear relationship (r>0.999) and a detection limit of 0.005 mg/kg. At the spiked levels of 0.005, 0.01 mg/kg and 0.02 mg/kg, the average recovery rates for DNOC were in the range of 82.6%-108% with a relative standard deviation (RSD) of less than 10% (n = 6). This method proved rapid, accurate and highly sensitive and can therefore be used for the determination of DNOC residue in animal-origin foods.
solid-phase extraction (SPE);ultrahigh performance liquid chromatography-tandem mass spectrometry (UPLCMS/MS);DNOC;animal-origin food;herbicide
TS207.5
A
1002-6630(2011)14-0268-05
2010-10-10
国家质检总局行标修订任务项目(2009B803r)
吴岩(1981—),男,工程师,硕士,研究方向为食品安全检测技术。E-mail:nkwuyan@163.com
*通信作者:康庆贺(1959—),男,研究员,学士,研究方向为食品安全检测技术。E-mail:mkangqh@126.com
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15 11:42:26
应用化工(2021年4期)2021-05-20 09:43:36
化工管理(2020年26期)2020-10-09 10:05:16
环境污染与防治(2020年7期)2020-07-27 07:31:38
广东茶业(2019年2期)2019-06-18 10:24:24
农药科学与管理(2019年12期)2019-05-20 09:33:26
山东化工(2019年2期)2019-02-21 09:29:32
中成药(2018年1期)2018-02-02 07:20:31
中国药业(2014年17期)2014-05-26 09:07:45
中国药业(2014年24期)2014-05-26 09:00:16
