
壮阳健威丸质量标准研究
2011-01-30 06:23方翠芬余潇苓
中成药 2011年6期
方翠芬, 陈 炜, 余潇苓, 祝 明
(1.浙江省食品药品检验所,浙江杭州310004;2.杭州胡庆余堂药业有限公司,浙江 杭州310016;3.浙江省中医院,浙江杭州310006)
壮阳健威丸是由人参、肉苁蓉、鹿茸等十六味药组成的复方制剂,具有补肾壮阳,生精益髓的作用,用于阳虚畏寒,腰膝酸痛,阳痿。该品种质量标准收载于《中华人民共和国卫生部药品标准》中药成方制剂第十册中(标准编号为:WS3-B-1923-95)[1],原标准仅收载了人参和制何首乌的薄层色谱鉴别和常规检查项,经文献查阅,尚未见有壮阳健威丸中定量测定方法的报道。为控制药品质量,保证临床用药的安全性和有效性,按照国家标准提高计划的要求,我们参考文献,建立了枸杞子[2]和甘草[3]的薄层色谱鉴别,修改了人参[4]和制何首乌[5-6]的薄层色谱鉴别,制订了主要药味肉苁蓉中松果菊苷的HPLC测定方法[7-9],建立的定性、定量方法均简便、准确、专属性强,可以有效地控制本品的质量。
1 仪器与试药
惠普1100高效液相色谱仪(惠普1100紫外检测器),Waters2695高效液相色谱仪(Waters2487紫外检测器),TU-1901紫外-可见分光光度计。
枸杞子对照药材,人参对照药材,甘草对照药材,2,3,5,4'-四羟基二苯乙烯-2-O-β-D-葡萄糖苷对照品,人参皂苷Rg1对照品、人参皂苷Rb1对照品、人参皂苷Re对照品,松果菊苷对照品(111670-200502,供定量测定用),均购自中国药品生物制品检定所。
壮阳健威丸样品由杭州胡庆余堂药业有限公司生产并提供,样品批号分别为080104、080105、080106。乙腈、甲醇为色谱纯(Merck),水为超纯水,其他试剂均为分析纯。
2 薄层色谱鉴别
2.1 枸杞子的薄层色谱鉴别 取本品3 g,粉碎,加乙醇30 mL,加热回流1 h,滤过,滤液蒸干,残渣加水5 mL,微热使溶解,离心,取上清液置于已处理好的聚酰胺柱(30~60目,内径1.5 cm,柱高10 cm,用水湿法装柱)上,用水50 mL洗脱,弃去水液,再用30%乙醇50 mL洗脱,收集洗脱液,继用50%乙醇洗脱,收集洗脱液,备用。取30%乙醇洗脱液,蒸干,残渣加无水乙醇1 mL使溶解,作为供试品溶液。另取枸杞子对照药材0.5 g,同法制成对照药材溶液。再取按处方比例及工艺制备的枸杞子阴性对照样品3 g,同法制成阴性对照溶液。吸取供试品溶液和阴性对照溶液各10 μL、对照药材溶液5 μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯-三氯甲烷-甲酸(5∶3∶2∶0.5)为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的蓝色荧光斑点。见图1。

图1 枸杞子TLC图谱
2.2 制何首乌的薄层色谱鉴别 取枸杞子薄层色谱鉴别项下的50%乙醇洗脱液,蒸干,残渣加无水乙醇0.5 mL使溶解,作为供试品溶液。另取制何首乌对照药材0.5 g,同法制成对照药材溶液。再取2,3,5,4'-四羟基二苯乙烯-2-O-β-D-葡萄糖苷对照品,加无水乙醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。再取按处方比例及工艺制备的制何首乌阴性对照样品3 g,同法制成阴性对照溶液。吸取供试品溶液和阴性对照溶液各5 μL、对照药材溶液和对照品溶液各2 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸(15∶0.7∶0.5)为展开剂,展开,取出,晾干,喷以磷钼酸硫酸溶液(取磷钼酸2 g,加水20 mL使溶解,再缓缓加入硫酸30 mL,摇匀),热风吹至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。见图2。

图2 制何首乌TLC图谱
2.3 人参的薄层色谱鉴别 取本品3 g,粉碎,加乙醇30 mL,加热回流1 h,滤过,滤液蒸干,残渣加水15 mL使溶解,用三氯甲烷15 mL振摇提取,弃去三氯甲烷液,水层用水饱15 mL , ,0.6钠溶液15 mL振摇提取,碱水层备用,正丁醇液蒸干,残渣加无水乙醇1 mL使溶解,作为供试品溶液。另取人参对照药材0.2 g,同法制成对照药材溶液。再取人参皂苷Rg1、人参皂苷Rb1、人参皂苷Re对照品,加无水乙醇制成每1 mL含1 mg的混合溶液,作为对照品溶液。再取按处方比例及工艺制备的人参阴性对照样品3 g,同法制成阴性对照溶液。吸取供试品溶液、对照药材溶液和阴性对照溶液各2 μL、对照品溶液1 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光及紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,分别显相同颜色的斑点或荧光斑点。见图3。
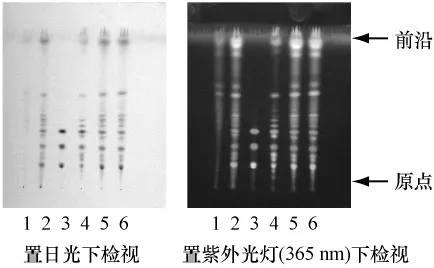
图3 人参TLC图谱
2.4 甘草的薄层色谱鉴别 取人参薄层色谱鉴别项下的备用碱水溶液,用盐酸调节pH值至2,加乙酸乙酯15 mL振摇提取,分取乙酸乙酯液,蒸干,残渣加无水乙醇1 mL使溶解,作为供试品溶液。另取甘草对照药材0.2 g,加乙醇30 mL,加热回流1 h,滤过,滤液蒸干,残渣加0.6%氢氧化钠溶液15 mL使溶解,用水饱和正丁醇15 mL振摇提取,分取碱液,自“用盐酸调节pH值至2……”起,同法制成对照药材溶液。再取按处方比例及工艺制备的甘草阴性对照样品3 g,同法制成阴性对照溶液。吸取供试品溶液和阴性对照溶液各5 μL、对照药材溶液2 μL,分别点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,分别置日光和紫外光灯(UV365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,分别显相同颜色的斑点或荧光斑点。见图4。

图4 甘草TLC图谱
3 松果菊苷的测定
3.1 色谱条件 色谱柱:ZORBAX SB C18(4.6 mm×250 mm,5 μm);柱温30℃;流动相乙腈-甲醇-0.1%甲酸溶液(7∶16∶77);体积流量1.0 mL/min;检测波长330 nm。
3.2 对照品溶液的制备 取松果菊苷对照品适量,精密称定,加50%甲醇溶解,制成每1 mL含松果菊苷28.9 μg的溶液,即得对照品溶液。
3.3 供试品溶液的制备 取壮阳健威丸样品,粉碎成细粉,取约1 g,精密称定,精密加50%甲醇25 mL,称定质量,超声处理(功率400 W,频率40 kHz)40 min,取出,放冷,再称定质量,用50%甲醇补足减失的质量,摇匀,离心,取上清液,即得。
3.4 阴性对照溶液的制备 取按处方比例及工艺制备的缺肉苁蓉样品,同供试品溶液制备方法制得阴性对照溶液。
在上述色谱条件下,精密吸取对照品溶液、供试品溶液和阴性对照溶液各20 μL,分别注入液相色谱仪测定,色谱图见图5,松果菊苷的保留时间为23.6 min,与相邻组分达到基线分离,阴性对照无干扰。理论板数按松果菊苷峰计算应不低于3 000。
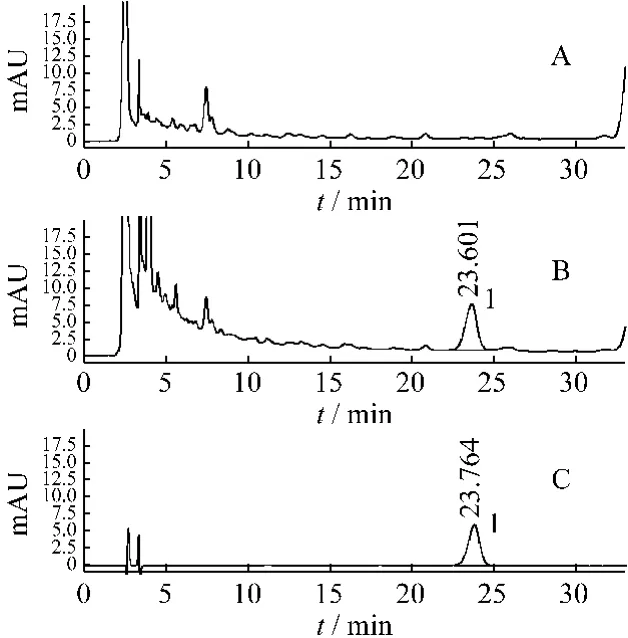
图5 HPLC色谱图
3.5 线性关系考察 精密吸取松果菊苷系列对照品溶液(质量浓度分别为 173.4、57.8、28.9、14.4、7.2、3.6、1.8 μg/mL)各20 μL,注入液相色谱仪,按上述条件测定峰面积,以峰面积为Y纵坐标,进样量为X横坐标进行线性回归,得回归方程:Y=1 314.4X-23.019;r=1.000 0,结果表明,松果菊苷在进样量0.036~3.468 μg范围内线性关系良好。
3.6 仪器精密度考察 精密吸取松果菊苷对照品溶液,按上述条件重复进样6次,测定峰面积,结果松果菊苷峰面积的RSD为1.0%(n=6)。表明仪器精密度良好。
3.7 重复性考察 取同一壮阳健威丸样品(批号:080104)6份,按3.3项下方法制备供试品溶液,按上述色谱条件测定峰面积,计算质量分数,结果平均质量分数为0.33 mg/g,RSD为0.7%(n=6)。
3.8 稳定性考察 取同一供试品溶液,室温下放置,按上述色谱条件,于 0、1、2、7、13、24、50 h 时测定,结果松果菊苷峰面积RSD为1.3%(n=7),表明供试品溶液中松果菊苷在50 h内基本稳定。
3.9 加样回收率试验 取已测定的壮阳健威丸样品(批号:080104)9份,精密加入1、2、3 mL松果菊苷对照品溶液(87.75 μg/mL),按3.3项下供试品溶液制备方法处理,测定,计算加样回收率,结果见表1。平均回收率为97.8%,RSD为1.2%。

表1 加样回收率实验结果
3.10 样品测定 按上述测定方法,对3批壮阳健威丸样品进行测定,结果见表2。

表2 样品测定结果
4 讨论
4.1 HPLC测定实验过程中,在200~400 nm波长范围内进行紫外光谱扫描,发现松果菊苷的最大吸收波长为332 nm,参考《中国药典》[10]2005年版一部“肉苁蓉”含量测定项下的测定波长为330 nm,考虑到紫外吸收波长扫描有一定的偏差,最终参考药典,选择330 nm为测定波长。
4.2 定量测定耐用性考察[11]实验中考察了柱温(25℃、30℃、35℃)对色谱分离的影响,结果发现柱温的改变对供试品溶液的分离效果产生较大影响,而当柱温在30℃时,分离情况较好,故建议柱温控制在30℃。实验中比较了3种品牌的色谱柱:ZORBAX SB-C18(4.6 mm ×250 mm,5 μm),Diamonsil C18(4.6 mm × 250 mm,5 μm),依利特 Hypersil ODS(4.6 mm×250 mm,5 μm),结果供试品在3种色谱柱上均能得到较好的分离。
[1]《中华人民共和国卫生部药品标准》中药成方制剂第十册[S].1995:53.
[2]苗明三,李振国.现代实用中药质量控制技术[M].北京:人民卫生出版社,2000:715.
[3]孙国英,张春丽,黄 超,等.椰露止咳合剂中鱼腥草和甘草薄层鉴别方法的改进[J].中药材,2009,32(7):1148-1150.
[4]莫结丽,隆 颖.救心丸中人参薄层色谱鉴别研究[J].中国医药导报,2009,6(4):38-39.
[5]谢 岚.何首乌及二苯乙烯苷的研究进展[J].天津药学,2010,22(3):70-73.
[6]高晓霞,严寒静,梁从庆.不同采集地制何首乌薄层色谱指纹图谱研究[J].中国实验方剂学杂志,2007,13(5):4-6.
[7]李 媛,宋媛媛,张洪泉.肉苁蓉的化学成分及药理作用研究进展[J].中国野生植物资源,2010,29(1):7-11.
[8]王哲民,王蔼英.HPLC测定苁蓉通便口服液中松果菊苷的含量[J].中成药,2006,28(7):1072-1073.
[9]刘 雄,吴 蓉,余晓晖,等.HPLC法测定不同产地肉苁蓉中松果菊苷的含量[J].甘肃中医学院学报,2009,26(5):42-44.
[10]国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:126.
[11]国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:附录130.
猜你喜欢
金沙江文艺(2022年1期)2022-02-04
化学教与学(2021年12期)2021-02-18
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24
数学小灵通(1-2年级)(2020年3期)2020-06-24
时代邮刊·下半月(2019年8期)2019-09-10
小学生导刊(2018年34期)2018-12-18
数学小灵通(1-2年级)(2018年4期)2018-05-07
海峡科技与产业(2016年3期)2016-05-17
云南中医学院学报(2015年3期)2015-07-31
小天使·二年级语数英综合(2015年5期)2015-05-15
