
利用RNA捕获法在全基因组进行egfr启动子片段的富集
2011-01-25 10:26:08邹斌斌张玉祥王泽生
首都医科大学学报 2011年2期
杨 锦 邹斌斌 张玉祥 王泽生
(首都医科大学基础医学院生物化学与分子生物学系)
表皮生长因子受体(epidermal growth factor receptor,EGFR)参与调控细胞代谢、增生、迁移和分化。其过度表达可加速细胞分裂与增生,抑制凋亡,有助于血管形成,并促进癌细胞转移[1]。人类肿瘤包括非小细胞肺癌、乳腺癌、头颈癌、胃癌、结肠癌、食管癌、前列腺癌、膀胱癌、肾癌、胰腺癌、口腔癌等都可表达或高表达EGFR[2]。肿瘤中的EGFR表达与疾病侵袭性增强、化疗抵抗增加、转移性增强,生存率下降及临床预后差相关[3]。
egfr基因5'调控区包括1个富含GC的启动子,缺少保守序列TATA盒和CAAT盒,有多个位点可以起始转录[4]。egfr启动子在-216G/T存在多态性并与其增加活性相关[5]。深入了解egfr基因在肿瘤内的表达调控将有助于我们对该基因的表达进行干预,并治疗相应肿瘤。进行这类研究需要大规模、多样本的基因组重测序。尽管第二代测序成本已明显降低,但全基因组重测序需要大量的时间和高额的成本,制约其应用于疾病研究。基因组目标区域重测序策略不需对全基因组进行测序,只对基因或其他感兴趣的基因组区域的基因序列重新测定。国际上已发表的研究报告[6-7]均采用商业公司推出的目标序列捕获解决方案。本研究以宫颈癌细胞系HeLa S3 egfr启动子为模型,建立了一套简便易行的RNA捕获靶序列的富集体系,结合Solexa测序方法,为各种复杂人类疾病相关突变的检测奠定良好基础。
1 材料和方法
1.1 实验材料
1)细胞株:宫颈癌细胞系HeLa S3(购于北京协和细胞库)。
2)主要试剂:RPMI 1640培养基(美国Gibco公司);胎牛血清(fetal bovine serum,FBS)(美国Hyclone公司);MboI、Klenow Fragment、Klenow Fragment(3'→5'exo-)、T7 RNA Polymerase、rNTP(美国NEB公司); T4 DNA连接酶、Ex Taq(日本Takara公司);Biotin-11-dUTP(立陶宛Fermentas公司);Biotin-16-UTP(美国Ambion公司);Dynabeads M-280 Streptavidin(美国Invitrogen公司);DNA胶回收试剂盒、PCR纯化试剂盒(德国Qiagen公司);引物合成单位(中国上海生工生物工程有限公司);高通量测序单位(北京六合华大基因科技股份有限公司深圳分公司)。
1.2 方法
1)细胞培养:在5%CO2,37℃的培养箱中,用含有10%FBS的RPMI 1640培养基对HeLa S3细胞进行培养。
2)DNA文库的制备:①细胞基因组的提取:8 000 g离心1 min,收集细胞,加入终浓度为0.5 μg/μL蛋白酶K,55℃消化过夜。消化16 h后再加入终浓度为0.2 μg/μL的RNase A,在37℃水浴30 min消化RNA,酚/氯仿抽提,乙醇沉淀,加入50 μL无菌水溶解。②MboⅠ酶切基因组:取基因组2 μg,50 μL体系中加入5 μL 10×NE Buffer2,加入5 U的限制性内切酶MboⅠ,在37℃下水浴,消化过夜。65℃水浴20 min灭活限制性内切酶活性。③DNA片段两端加接头:加入2 μL 10× NE Buffer2,加入终浓度为0.1 mmol/L的dNTP,加入Klenow Fragment(3'→5'exo-)15 U,在37℃水浴30 min。将体系扩大到100 μL,加入终浓度为1×T4 DNA连接酶缓冲液,加入终浓度为0.8 μmol/L的3'端‘T’突出的Pair-end adapter 1和Pair-end adapter 2(表1),加入50 U的T4 DNA连接酶,16℃水浴过夜。④DNA片段的纯化、回收:酚/氯仿抽提,乙醇沉淀,加入15 μL无菌水溶解。所得DNA用1.5%琼脂糖胶电泳,QIAGEN试剂盒胶回收400~1 000 bp DNA,加入100 μL无菌水溶解作为杂交模板。
3)生物素标记egfr启动子片段的RNA制备和RNA/DNA杂交捕获:①egfr启动子片段两端加T7,SP6 adapter:以基因组DNA为模板,用egfr promotor primer (表1)扩增egfr启动子片段272 bp,液体回收后加上T7,SP6 adapter(表1),用T7,SP6引物PCR扩增egfr启动子片段,即得到带有T7启动子的egfr启动子片段。②egfr启动子片段转录为RNA:液体回收PCR产物,30 μL反应体系中含有4 mmol/L rNTP,1×T7 RNA Polymerase buffer,75 U T7 RNA Polymerase,0.3 mmol/L biotin-16-UTP,37℃水浴3 h。③egfr启动子RNA/3C模板杂交: 200 μL反应体系中含有5×SSC buffer(0.75 mol/L NaCl,0.075 mol/L柠檬酸钠),加入DNA模板100 μL,95℃加热2 min,冰上1 min,加入RNA 30 μL,65℃杂交24 h。④egfr启动子片段富集:取链亲和素包被的Dynal磁珠10 μL,用100 μL 2×B&W缓冲液悬浮磁珠,加入100 μL的生物素化RNA/DNA,轻轻旋转,室温下放置过夜。用200 μL 1×TWB缓冲液洗3次,70℃加热30 min,取上清,即得到生物素化的RNA/DNA(图1)。
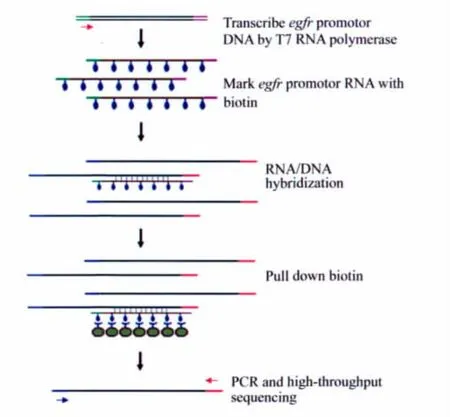
图1 RNA捕获法技术流程图Fig.1 Schematic representation of RNA capture method

表1 PCR引物及接头序列Tab.1 Sequence of pair-end adapters and PCR primers
4)样本扩增和高通量测序:50 μL反应体系中含有0.4 mmol/L PE引物(表1),0.2 mmol/L dNTPs,1.25 U Ex Taq DNA聚合酶,5 μL DNA模板。98℃变性30 s后,再依下列条件进行PCR:98℃变性10 s,62℃退火30 s,72℃延伸60 s,15个循环后,72℃延伸5 min。产物纯化定量。RNA捕获的样本扩增15个循环后,进行Solexa高通量测序。
2 结果
2.1 MboⅠ酶切基因组DNA
结果显示,MboⅠ酶消化的基因组DNA产生从大到小均一的酶切片段,与对照基因组相比,消化比较完全(图2)。
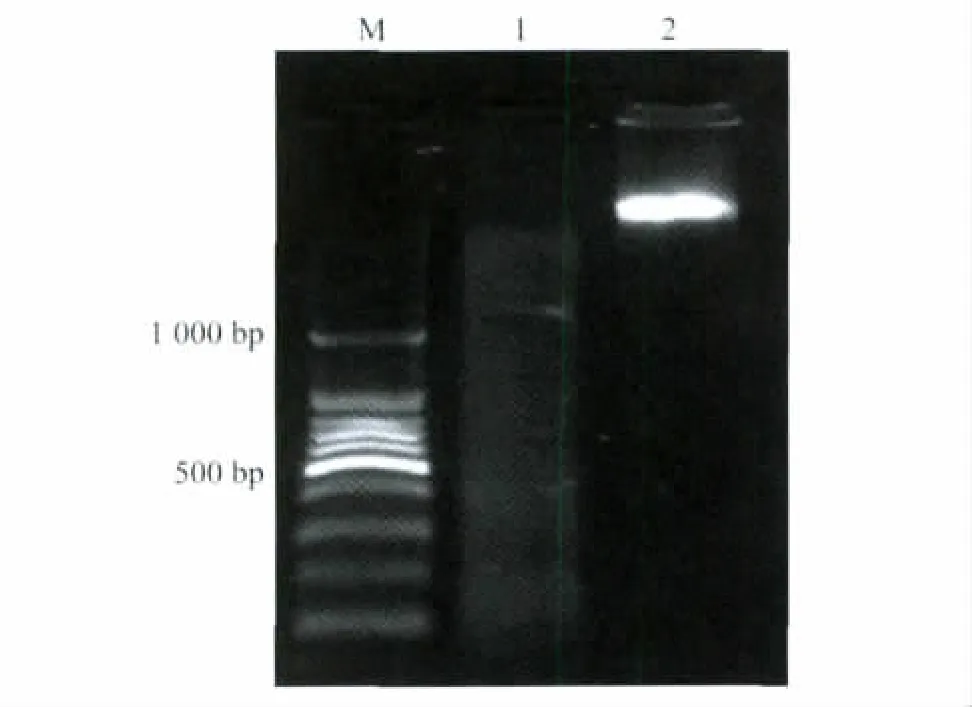
图2 MboⅠ酶切基因组DNA琼脂糖凝胶电泳Fig.2 MboⅠdigestion efficiency by gelelectrophoresis assay M:100 bp DNA ladder;1:HeLa S3 genome DNA treated by MboⅠ;2:HeLa S3 genome DNA untreated by MboⅠ with same quantity as control.
2.2 egfr启动子片段转录为RNA
用T7 RNA Polymerase转录egfr启动子272 bp,37℃水浴3 h后,用1.5%琼脂糖凝胶电泳检测转录产物,可见egfr启动子RNA呈弥散分布(图3)。
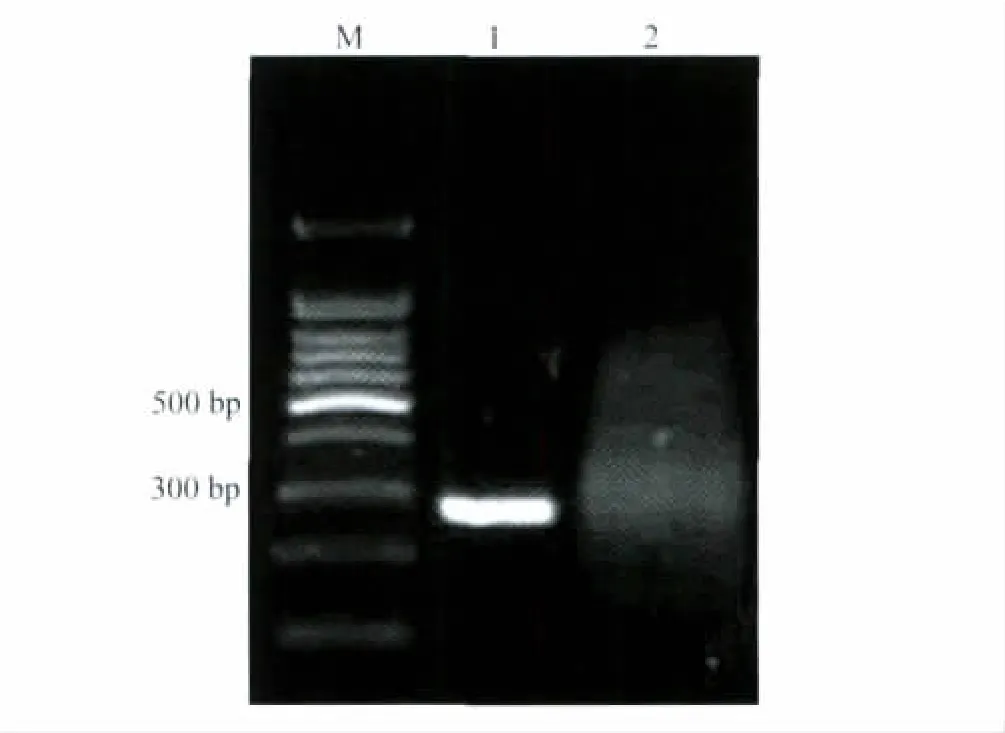
图3 T7 RNA Polymerase转录egfr启动子片段琼脂糖凝胶电泳Fig.3 Agarose gelelectrophoresis of egfr promotor DNA transcription by T7 RNA PolymeraseM:100 bp DNA ladder;1:egfr gene promotor DNA;2:egfr promotor RNA after transcription.
2.3 egfr启动子片段富集的验证
分别取富集前和富集扩增后的DNA模板12 ng作模板,用egfr promotor primer(表1)扩增,产物液体回收纯化。琼脂糖胶电泳结果显示egfr启动子片段经过RNA杂交捕获后有富集(图4)。
2.4 Solexa高通量测序结果评价:测序结果Reads比对不同的基因分析
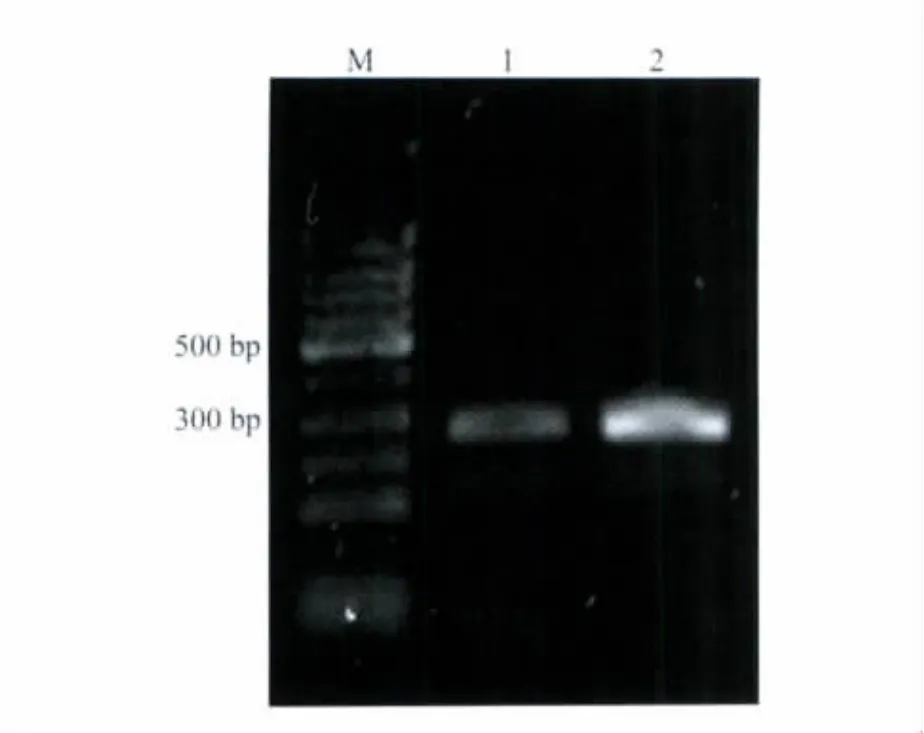
图4 egfr启动子片段PCR琼脂糖凝胶电泳Fig.4 Agarose gelelectrophoresis of egfr gene promotor DNA PCRM:100 bp DNA ladder;1:egfr gene promotor PCR using template before enrichment;2:egfr gene promotor PCR using template after enrichment.
Solexa高通量测序结果的数据作为实验组,用黑色表示,从实验组中随机取总条数为106reads,比对到egfr启动子片段reads条数为1 889条,而比对到随机挑选的notch 1、pdgf-B、src基因reads条数为2、3、3条。从人类全基因组随机取总条数106的等长reads作为对照组,用白色表示,比对到notch 1、pdgf-B、src、egfr基因reads条数分别为3,2,3,2条。横坐标轴表示各基因名称,纵坐标轴表示比对到各基因片段reads条数取log值。分析显示,RNA捕获法在本实验中进行全基因组egfr启动子片段富集了630倍(图5)。
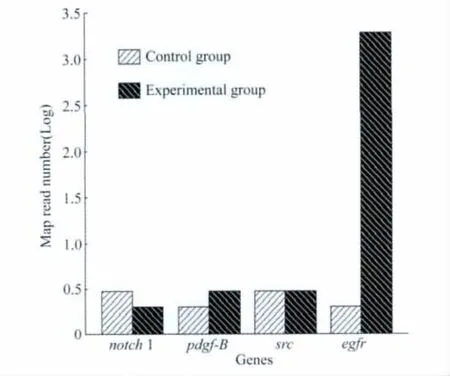
图5 Reads比对不同的基因分析Fig.5 Analysis of Reads mapping different genes
3 讨论
基因组测序分析能够为基因DNA序列提供最真实可靠的信息,它可以比较全面地描述基因的复杂性和多样性[8]。随着人类基因组计划的完成和测序技术的不断发展,可以大规模地筛选突变基因,并对基因多态性进行检测,有助于发现罕见的基因突变和肿瘤组织中基因组水平的变异[9]。但是在全基因组水平进行测序目前的费用还比较昂贵,一般课题组难以承受,基因诊断和治疗技术的普及受到DNA富集方法的限制。本课题组采用RNA捕获法将egfr启动子片段转录为RNA,与基因组进行杂交富集,并洗去基因组的其他部分,用PCR扩增建立测序文库,进行高通量测序并利用生物信息学技术进行分析,证明RNA捕获法对egfr启动子有明显的富集作用。传统的靶向重测序技术如PCR技术,费用过高或者通量过低[6],该技术与之相比,具有一定的优越性,它不存在偏向性,可以高效富集DNA,针对性强,简便易行,且降低了测序成本。Nimbl Gen公司[10]推出的基于芯片杂交的全外显子捕获芯片,内含210万个寡核苷酸探针,可以捕获人类大部分外显子,通过测序识别在外显子中的基因变异。Agilent公司[11]推出了基于芯片杂交和液相杂交的全基因组外显子捕获产品,捕获探针的长度是120 bp,每管溶液中包含55 000个生物素化的RNA探针,Agilent公司还发展了在线eArray设计平台,可以根据客户需要自行设计探针。但是对于一般的研究单位而言,费用仍较高,需要提前订购,在技术也并非简便易行。
本技术利用RNA捕获法,不仅可以对感兴趣的靶区段自行设计,还可以自行制备生物素化的RNA探针,降低了成本。除了对外显子或编码序列设计探针外,还可以对感兴趣的非编码序列自行设计和制备RNA探针。本实验RNA探针的长度达272 bp,而且在序列捕获时采用较高温度,提高了捕获反应的特异性。本技术结合高通量技术深度测序所富集区域,而非全基因组序列,从而提高效率,降低成本。它可以用于大规模、多样本的与疾病相关的基因和基因组水平异常的研究,包括单核苷酸多态性,基因组结构变异,群体多态性,可变剪切等。该技术为研究人员提供了一种低成本、高效率、灵活的目标测序方法,可以适用于多种不同的应用领域,它的研究与发展将会在重大疾病研究中发挥重要作用。
[1] Ciardiello F,Tortora G.EGFR antagonists in cancer treat-ment[J].N Engl J Med,2008,358:1160-1174.
[2] Salomon D S,Brandt R,Ciardiello F,et al.Epidermal growth factor-related peptides and their receptors in human malignancies[J].Crit Rev Oncol Hematol,1995,19:183-232.
[3] Tabernero J.The role of VEGF and EGFR inhibition:implications for combining anti-VEGF and anti-EGFR agents[J].Mol Cancer Res,2007,5:203-220.
[4] Brandt B,Meyer-Staeckling S,Schmidt H,et al.Mechanisms of EGFR gene transcription modulation:relationship to cancer risk and therapy response[J].Clin Cancer Res,2006,12:7252-7260.
[5] Liu W,Wu X,Zhang W,et al.Relationship of EGFR mutations,expression,amplification,and polymorphisms to epidermal growth factor receptor inhibitors in the NCI60 cell lines[J].Clin Cancer Res,2007,13:6788-6795.
[6] Mamanova L,Coffey A J,Scott C E,et al.Target-enrichment strategies for next-generation sequencing[J].Nat Methods,2010,7:111-118.
[7] Gnirke A,Melnikov A,Maguire J,et al.Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing[J].Nat Biotechnol,2009,27: 182-189.
[8] Shendure J,Ji H.Next-generation DNA sequencing[J].Nat Biotechnol,2008,26:1135-1145.
[9] Hillier L W,Marth G T,Quinlan A R,et al.Whole-genome sequencing and variant discovery in C.elegans[J].Nat Methods,2008,5:183-188.
[10]Choi M,Scholl U l,Ji W,et al.Genetic diagnosis by whole exome capture and massively parallel DNA sequencing[J].Proc Natl Acad Sci USA,2009,106:19096-19101.
[11]Metzker M L.Sequencing technologies——the next generation[J].Nat Rev Genet,2010,11:31-46.
猜你喜欢
太原理工大学学报(2021年6期)2021-11-25 13:33:20
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21 07:26:06
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04 16:33:12
畅谈(2018年6期)2018-08-28 02:23:38
数学小灵通(1-2年级)(2017年12期)2018-01-23 03:37:05
计算机测量与控制(2017年6期)2017-07-01 16:24:07
广州化工(2016年11期)2016-09-02 00:42:59
非公有制企业党建(2016年1期)2016-07-19 13:02:52
开心素质教育(2016年2期)2016-04-20 05:07:48
读写算·小学低年级(2015年4期)2015-12-04 11:27:22
