
乙氧苯柳胺软膏工艺改变前后透皮实验研究
2011-01-24 09:34刘建军钟巧妮
医药导报 2011年11期
刘建军,钟巧妮
(湖北省医药工业研究院有限公司,武汉 430061)
笔者在本实验中考察乙氧苯柳胺软膏工艺改变前后的透皮效果,将乙氧苯柳胺工艺改变前后的两批样品分别涂于鼠皮,利用40%乙醇/pH7.4的磷酸溶液作为接收介质,通过不同时间点取样,采用高效液相色谱法测定接收介质中乙氧苯柳胺含量,通过各时间点乙氧苯柳胺累积透过皮肤量比较工艺改变前后是否一致[1-2]。
1 仪器与试药
透皮仪TT-6,Agilent1200高效液相色谱仪,色谱柱:ODS 柱(150 mm×4.6 mm,5μm),流动相:甲醇∶水(67∶33)用磷酸调 pH 至 2.6,流速:1.0 mL·min-1,检测波长:UV290 nm,柱温:室温,进样量:20 μL。工作对照品(标准品,含量:99.8%,山东新华制药股份有限公司提供),乙氧苯柳胺软膏样品(规格:每支10 g,山东新华制药股份有限公司提供,批号:0910003,0911015),甲醇为色谱纯,其余试剂均为分析纯。
2 实验方法
2.1 鼠皮的制备 取健康雄性大鼠,剃净腹部毛,断颈处死,立即剥离皮肤,去净皮下脂肪,用0.9%氯化钠溶液反复冲洗,剪成适宜大小,浸于0.9%氯化钠溶液中,放入冰箱中-20℃保存待用,于一周内用完。每次实验前目测检查鼠皮完整性,不得有任何破损。
2.2 溶液的制备
2.2.1 标准品溶液的制备 精密称取乙氧苯柳胺10.01 mg(含量:99.8%),置 200 mL 容量瓶,接收介质溶解并稀释至刻度,摇匀,得49.9 μg·mL-1标准储备液。
2.2.2 供试品溶液的制备 将不同处方的乙氧苯柳胺软膏各0.25 g分别均匀涂布于皮肤角质层上。接受池中加入40%乙醇/pH7.4磷酸盐缓冲液12.0 mL作为接受介质,37℃恒温水浴,释放面积为1.77 cm2。定速搅拌,于2,4,6,8,10,22 h 取接受液1.0 mL,同时向接受池中加入等量同温的空白接收介质。将所取接受液经微孔滤膜过滤,作为供试品溶液。
2.2.3 阴性对照溶液的制备 取准备好的鼠皮两张,采用透皮仪进行实验[3],将皮肤分别置于接收池和供给池之间,角质层面向上,其中一张鼠皮作为空白,另一张皮肤上涂不含乙氧苯柳胺的软膏基质,作为含基质的空白。然后在接受池中加入40%乙醇/pH7.4磷酸盐缓冲液12.0 mL作为接受介质,37℃恒温水浴,释放面积1.77 cm2。定速搅拌,24 h后取接受液3 mL,将所取接受液经微孔滤膜过滤,作为空白溶液。
2.3 实验方法与结果
2.3.1 专属性实验 分别取标准品溶液、阴性对照溶液进样检测,结果空白液在标准品主峰处均无吸收峰出现。
2.3.2 线性关系考察 精密吸取“2.2.1”项下储备液,依次稀释成浓度为 0.100,0.499,0.999,4.995,9.990和 19.980 μg·mL-1的标准溶液,取 20 μL 进样,记录峰面积。以浓度(C)对峰面积(A)进行线性回归,得线性方程:Y=76.309X-6.667 3,结果可见本品在 0.100 ~19.980 μg·mL-1范围内线性关系良好,r=0.999 8。
2.3.3 精密度实验 取“2.3.2”项下4.995 μg·mL-1的乙氧苯柳胺标准溶液,重复测定6次。结果峰面积分别为 366.939,367.335,367.175,367.448,367.245,367.008,平均 367.192,RSD=0.05%。可见本品精密度较好。
2.3.4 回收率实验 取辅料基质0.203 2 g,置于100 mL量瓶,加接收介质液溶解,并稀释至刻度,作为辅料储备液;分别精密吸取线性项下49.9 μg·mL-1标准储备液和辅料储备液,配制线性浓度范围内高、中、低浓度的乙氧苯柳胺样品溶液各3份,高效液相色谱法测定,由标准曲线回归方程计算其浓度测定值,测定值与真实值的比值得回收率,结果见表1,平均回收率99.45%,RSD=1.0%。可见本品回收率较高。

表1 乙氧苯柳胺回收率实验结果
3 体外透皮释药研究
3.1 实验条件 接收介质:笔者对pH7.4磷酸缓冲液、10%乙醇/pH7.4磷酸缓冲液、20%乙醇/pH7.4磷酸缓冲液、30%乙醇/pH7.4磷酸缓冲液、40%乙醇/pH7.4磷酸缓冲液、30%丙二醇/pH7.4磷酸缓冲液等不同接收介质进行了溶解性对比。结果显示,样品在前3种接收介质条件下较难溶解,在30%乙醇/pH7.4磷酸缓冲液和30%丙二醇/pH7.4磷酸缓冲液中溶解较好,在40%乙醇/pH7.4磷酸缓冲液溶解最好。因此,最后确定以在40%乙醇/pH7.4磷酸缓冲液作为接收介质进行实验。
3.2 实验方法 取不同处方的乙氧苯柳胺软膏,按“2.2.2”项方法操作,取供试液 20 μL,按高效液相色谱法测定,由“2.3.2”项下标准曲线计算药物浓度。
3.3 数据处理与结果 经高效液相色谱法测得的药物浓度根据公式②进行校正,经公式③计算药物的累n-1Ci积渗透量。②Cn'=Cn+V/Vo∑t-1;③Qn=Cn‘*Vo/A。式中Cn‘:t时间药物的累积浓度,Ci:t时间前药物的测定浓度,Cn:t时间药物的测定浓度;V:取样体积;Vo:接受池中溶液的体积;Qn:t时间单位面积累积渗透量;A:皮肤有效扩散面积。测定每一取样点样品的浓度,计算单位面积累积渗透量(Q),结果见表2。以Qt作图,见图1。透皮速率见表3,方差分析见表4。
表2 改变工艺前后不同时点透皮实验结果
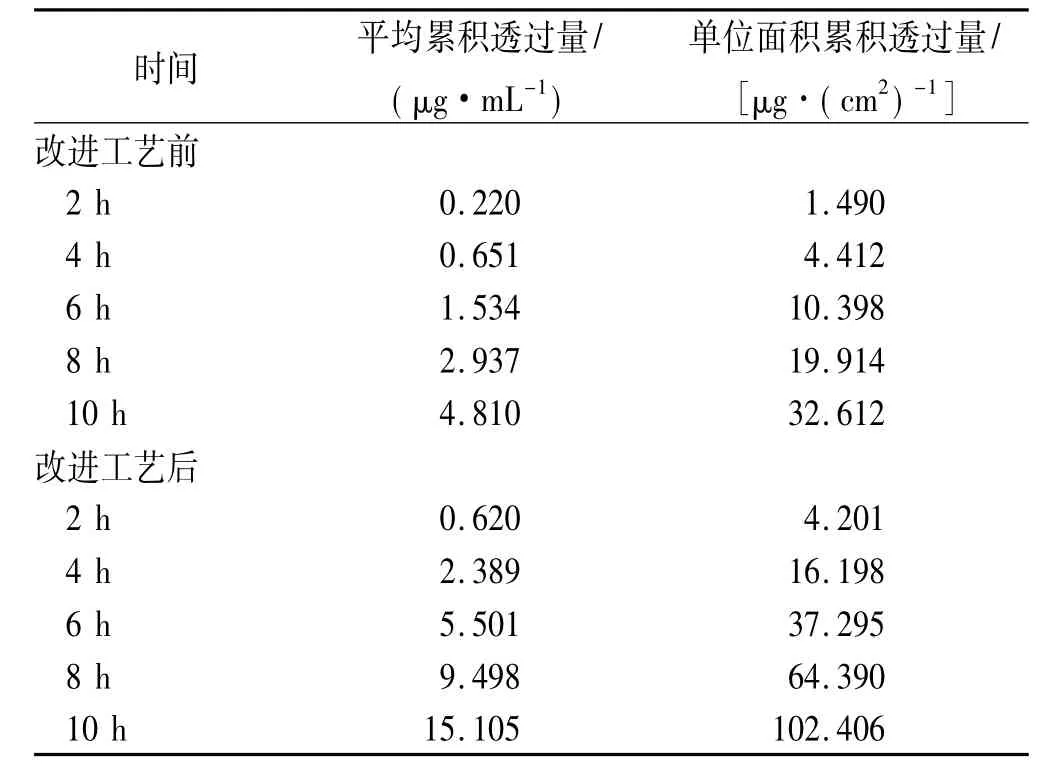
表2 改变工艺前后不同时点透皮实验结果
时间 平均累积透过量/(μg·mL-1)单位面积累积透过量/[μg·(cm2)-1]改进工艺前2 h 0.220 1.490 4 h 0.651 4.412 6 h 1.534 10.398 8 h 2.937 19.914 10 h 4.810 32.612改进工艺后2 h 0.620 4.201 4 h 2.389 16.198 6 h 5.501 37.295 8 h 9.498 64.390 10 h 15.105 102.406
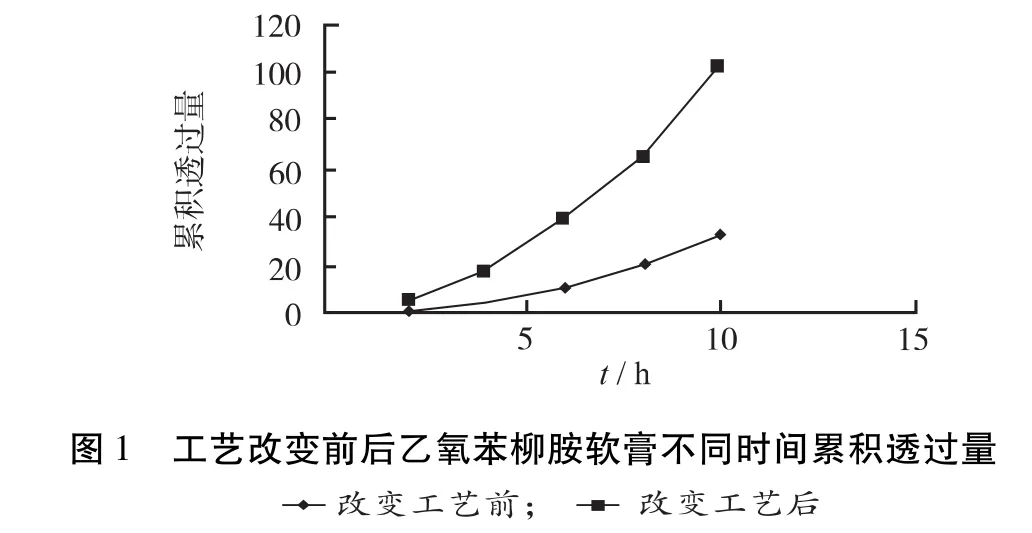

表3 乙氧苯柳胺软膏改变工艺前后透皮速率与10 h累积渗透量
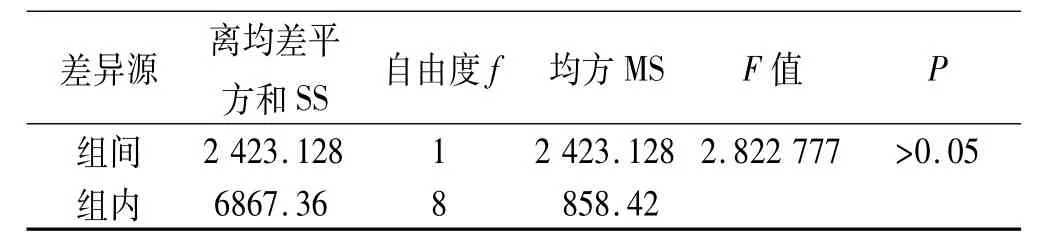
表4 透过量进行方差分析
4 结论
因22 h取样测定结果超出线性范围未纳入统计。从表3,4中可以看出,10 h内工艺改变前、后乙氧苯柳胺的总透过量仅为涂抹量的0.5%和1%。表3数据说明样品在强透皮接收介质下,工艺改变后的渗透速率略高于工艺改变前;但表4分析结果显示P>0.05,说明工艺改变前后样品的透皮现象差异无统计学意义,表明两者的治疗效果可能相当[4]。
[1]乙氧苯柳胺软膏质量标准WS-009(X-007)-97-(1)新药转正标准54册[S].209.
[2]曲秀梅,吴建辉.反相HPLC法测定乙氧苯柳胺软膏的含量[J].黑龙江医药科学,2002,25(5):82.
[3]关玉晶,部琪臻,姜建华,等.自乳化基质乙氧苯柳乳膏的研制及透皮研究[J].中国新药杂志,2006,15(20):1766-1768.
[4]王波,张桂芳.HPLC法测定乙氧苯柳胺软膏的含量[J].药物分析杂志,2002,22(1):72-73.
猜你喜欢
磷肥与复肥(2022年6期)2022-08-02
云南化工(2022年4期)2022-05-17
能源工程(2021年6期)2022-01-06
临床与实验病理学杂志(2021年10期)2021-12-13
商品与质量(2021年31期)2021-08-20
中国科技纵横(2021年24期)2021-03-02
恋爱婚姻家庭·养生版(2020年7期)2020-08-10
恋爱婚姻家庭(2020年21期)2020-07-23
科学导报·学术(2020年19期)2020-07-09
自我保健(2020年12期)2020-02-06
