
高能量密度材料3,3′-偶氮-1,2,4,5-四嗪衍生物的分子设计
2010-11-30 10:49张婧婧高洪伟王朝杰
物理化学学报 2010年12期
张婧婧 高洪伟 卫 涛,* 王朝杰
(1温州医学院仁济学院,浙江温州 325035; 2温州医学院药学院,浙江温州 325035)
高能量密度材料3,3′-偶氮-1,2,4,5-四嗪衍生物的分子设计
张婧婧1高洪伟2卫 涛2,*王朝杰2
(1温州医学院仁济学院,浙江温州 325035;2温州医学院药学院,浙江温州 325035)
运用密度泛函理论(DFT)方法,计算系列3,3′-偶氮-1,2,4,5-四嗪衍生物的生成热.结果显示:—N3取代基在增加3,3′-偶氮-1,2,4,5-四嗪衍生物的生成热方面起了非常重要的作用.通过分析标题化合物的最弱键离解能发现:—NH2或—N3取代基非常有利于增加衍生物的热稳定性.计算的爆速(D)和爆压(p)数值表明:—NO2或—NF2取代基有利于提高3,3′-偶氮-1,2,4,5-四嗪衍生物的爆轰性能.综合爆轰性能和热稳定性的计算结果,3种3,3′-偶氮-1,2,4,5-四嗪衍生物可以作为潜在的品优高能量密度材料(HEDM)候选物.
密度泛函理论;3,3′-偶氮-1,2,4,5-四嗪;生成热;键离解能;爆轰性能
六元杂氮环成为传统含能材料的基本单元,特别是S-四嗪环(1,2,4,5-四嗪环).近些年,S-四嗪衍生物作为高能量密度材料(HEDM)无论在理论还是在实验合成方面都得到了广泛关注[1-7],归因于它们具有高密度、高正生成热以及较好的热稳定性等特点.S-四嗪衍生物中[3,3′-偶氮-(6-氨基-1,2,4,5-四嗪) (DAAT)]以及类似衍生物的合成和性能研究已有相关报道[8-11],具有良好的应用前景,一些可作为潜在的高爆炸物、火箭推进剂以及烟火型气体发生剂原料等.尽管不含氧,但DAAT展现出了较好的爆轰性能、高正生成热以及感度低、稳定性好等特点.DAAT预测生成热高达1000 kJ·mol-1[8],实验测量值在862-1034 kJ·mol-1[9-10]之间,实验测量爆速为7.40 km·s-1(ρ=1.64 g·cm-3)、爆压为24.00 GPa(ρ=1.61 g· cm-3)[12].Wilcox等[8]对DAAT以及含有—NO2取代3, 3′-偶氮-1,2,4,5-四嗪衍生物的生成热和分子结构进行了理论研究.虽然DAAT已经表现出较好的综合特性,但同时也存在密度较低和氧平衡偏低等不足.尽管DAAT已经成功合成并已有一些相关报道,但一些爆轰性能以及热力学性能仍然缺乏,另外其它类似衍生物也尚未合成.因此,迫切需要对DAAT及其类似衍生物的设计和合成进一步研究,寻找具有高爆轰性能、较高稳定性和高密度的品优3,3′-偶氮-1,2,4,5-四嗪类HEDMs.
由于一些化合物的合成较为困难且具有一定危险性,因此在设计HEDM方面,理论预测就成为行之有效的方法.理论研究不仅为筛选候选化合物提供了可能性,而且可以揭示其结构与性能之间的关系,因此可以为实验设计提供更多、更有效的基础性数据[13-14].本文报道了系列3,3′-偶氮-1,2,4,5-四嗪衍生物的气相生成热、热稳定性以及含能性质.首先运用密度泛函理论(DFT)[15-18],通过设计的等键反应预测标题化合物的生成热,然后利用键离解能评估了化合物的热稳定性,最后在计算的生成热和密度基础上预测了它们的爆速和爆压.希望这些结果能为设计理想的HEDM提供必要信息,并且能够对实验合成以及性能研究起到实际的指导意义.
1 计算方法
运用DFT方法结合设计的等键反应,在B3LYP/ 6-311G**和B3P86/6-311G**水平上预测了标题化合物的生成热.先前的报道[4]表明6-311G**基组的计算结果能够较好地符合实验值.基于总能量计算的等键反应方法已成功应用于预测化合物的生成热[425].在等键反应中,因反应物和生成物电子环境相近,由电子相关能造成的误差可以相互抵消,使求得的生成热误差大为降低[23].在设计的等键反应中1,2,4,5-四嗪的基本环骨架未破坏,选取1,2,4,5-四嗪和CH3N=NCH3为参考物质,降低了计算误差,该方法已被证明非常可靠[4,5,1921].目标化合物被分成3组(A、B和C三个系列),见图1.先前的工作已初步判定带有—NO2的衍生物其性能较为理想[4-5],因此C系列衍生物一端取代基定为—NO2,用以探讨当另一侧引入其它基团后对其结构以及性能的影响.
在298 K下,用于计算标题化合物生成热的等键反应设计如下:
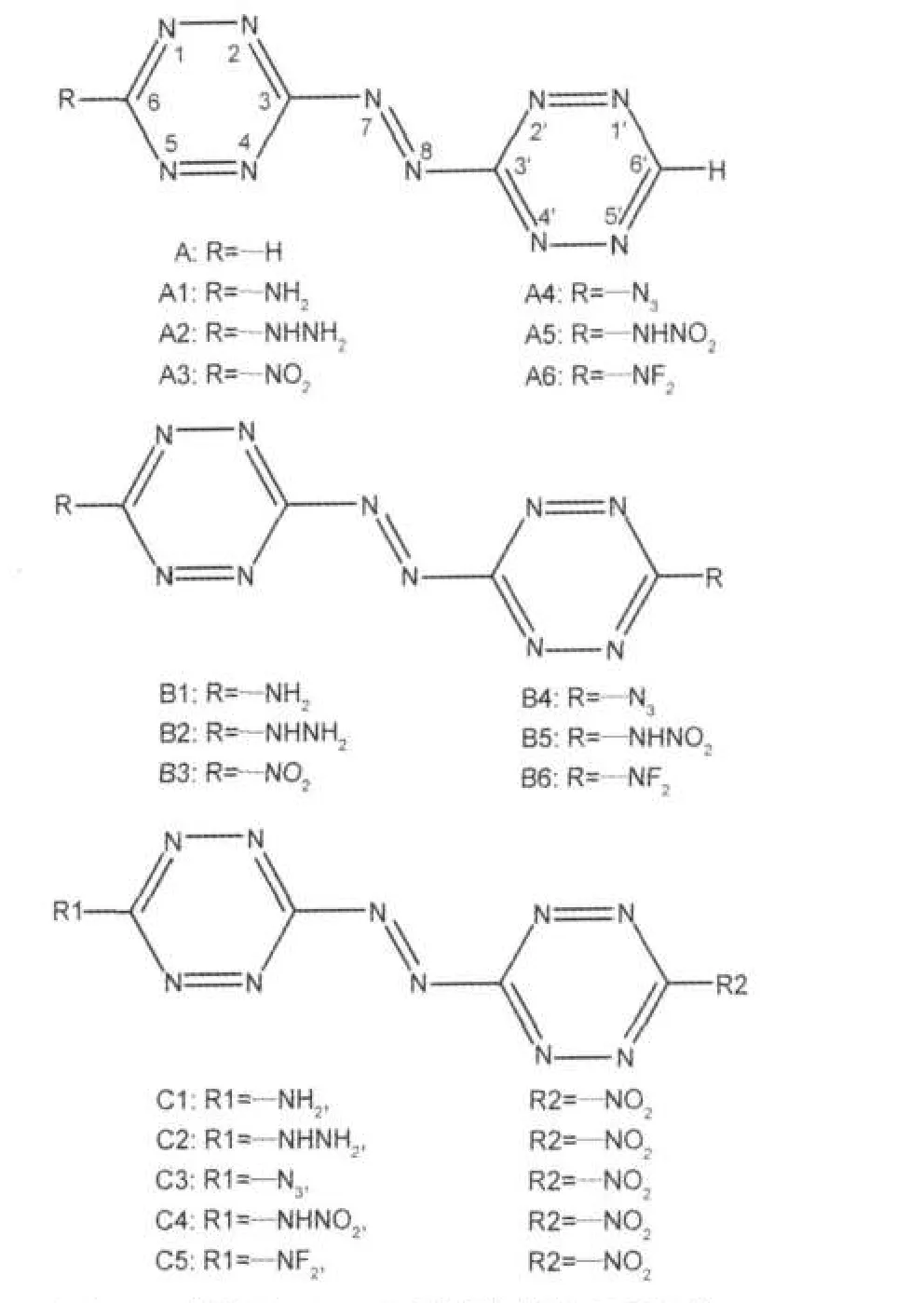
图1 3,3′-偶氮-1,2,4,5-四嗪衍生物的分子结构Fig.1 Molecular frameworks of 3,3′-azobis-1,2,4,5-tetrazine derivatives

其中R(或R1,R2)=—NH2、—NHNH2、—NO2、—N3、—NHNO2和—NF2.X表示3,3′-偶氮-1,2,4,5-四嗪, Tz表示1,2,4,5-四嗪.对于等键反应(1)和(2),在298 K时的反应热(ΔH298K)可表示如下:

式中ΔHf,r和ΔHf,p分别表示反应物和生成物在298 K时的生成热.参考物质Tz、CH4、CH3N=NCH3、CH3NH2、CH3NHNH2和CH3NO2的气相生成热是已知的[26].由于CH3N3、CH3NHNO2和CH3NF2缺乏实验值,因此需要借助于G2方法计算精确生成热[27-28].通过设计原子化反应(4)和(5),可以精确计算CH3N3和CH3NF2的生成热,而CH3NHNO2生成热的计算基于等键反应(6).

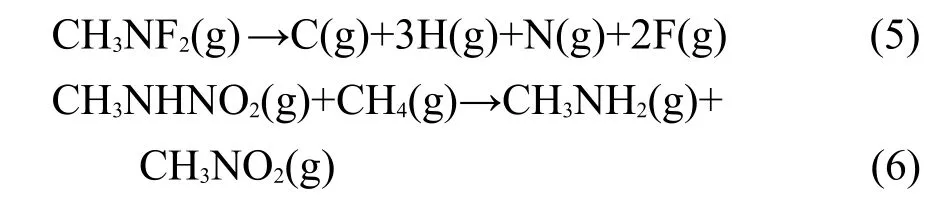
当已知反应热ΔH298K时,则可按照式(3)求得标题化合物的生成热.ΔH298K可以按照如下公式进行计算:

式中ΔE0表示0 K时产物与反应物的总能量之差, ΔZPE为0 K时产物与反应物的零点能(ZPE)之差; ΔHT是0-298 K的温度校正项.在理想气体反应中,Δ(pV)=ΔnRT.对于等键反应(1)和(2),Δn=0,因此Δ(pV)=0.
化学键的强度可用键离解能(BDE)来评价,它对于了解化学反应过程十分重要[29].在UB3LYP/ 6-311G**水平上计算标题化合物的键离解能,用以对比化合物的键强度以及热稳定性.在298 K和101.325 kPa下化学键均裂的能量等于反应A—B(g)→A·(g)+B·(g)的反应焓,该反应焓可以定义为A—B的键离解焓[30].文献报道许多有机化合物的键离解能(BDE)和键离解焓通常是近似相等的[31].因此,在0 K下物质的键离解能可以依据式(8)进行计算[31]:
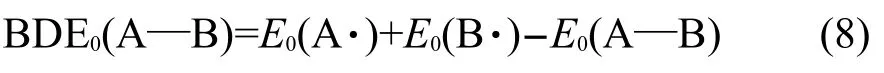
运用式(9)可以计算零点能(ZPE)校正的键离解能:

其中ΔZPE表示生成物与反应物的零点能之差.
运用Kamlet和Jacobs于1968年提出的半经验K-J方程[32]估算其爆速(D)、爆压(p)值:


式中p为爆压(GPa),D为爆速(km·s-1),ρ为密度(g· cm-3),N为每克炸药爆轰生成气体的摩尔数(mol· g-1),为气体产物的平均摩尔质量(g·mol-1),Q为每克炸药的爆轰化学能(J·g-1),结合等键反应所得生成热后,参照文献[32]的方法求得Q.计算化合物的密度必须已知摩尔体积,基于0.001 e·bohr-3的等电子密度面所包围的体积空间,应用Monte Carlo方法求算摩尔体积,这种方法已经成功应用于高氮化合物[33].对标题化合物在B3LYP/6-31G*理论水平上进行几何优化获得稳定构型,对每一个优化稳定构型执行100次单点计算取平均值.
计算方法为B3LYP/6-311G**和B3P86/6-311G**,所有计算使用Gaussian 09量子化学软件包[34]完成.收敛精度取程序默认值,振动分析表明所有得到的优化构型在此计算水平上没有虚频率,都是势能面上的局域极小点.
2 结果与讨论
2.1 生成热
生成热是化合物的基本热力学性质,是其含能高低的标志,也是衡量HEDM爆轰性能的重要参数,因此精确计算生成热对设计、筛选以及合成新型品优HEDM具有重要意义.一些高氮化合物标准生成热数据仍然缺少文献值或不易查找,其标准生成热的估算无论在理论上还是在实验中均具有重要意义.本文通过设计等键反应对标题化合物的生成热进行了计算.图1给出了3,3′-偶氮-1,2,4,5-四嗪衍生物的分子结构.表1列出等键反应(1)和(2)中9种参考物质的在B3LYP/6-311G**和B3P86/6-311G**水平上计算的总能量、零点能和温度校正项.对振动频率以校正因子0.96进行校正(参考Scott和Radom的研究结果[35]).在表1中参考物质的气相实验生成热参考文献[26](包括Tz、CH4、CH3N=NCH3、CH3NH2、CH3NHNH2和CH3NO2).CH3NF2和CH3N3的精确生成热分别参考文献计算值[22,25].另外,在G2水平上运用式(7)计算了反应(6)的等键反应反应热(ΔH298K)[2728]. CH4(g)、CH3NH2(g)和CH3NO2(g)的生成热实验值是已知的[26],故可以计算CH3NHNO2的精确生成热(ΔHf).为了验证理论计算的可靠性,我们在G2水平上计算了NH2NO2的生成热.结果显示,通过设计等键反应:NH2NO2(g)+CH4(g)→NH3(g)+CH3NO2(g), G2方法计算的生成热为-3.20 kJ·mol-1,与实验值(-3.20 kJ·mol-1[20])完全一致.由于CH3NHNO2和NH2NO2具有相似的结构特点,说明G2方法计算的CH3NHNO2生成热应该是可靠的.

表1 计算的参考物质总能量(E0)、零点能(ZPE)、温度校正项(HT)和生成热(HOFs)aTable 1 Calculated total energies(E0),zero-point energies(ZPE),thermal corrections(HT),and heats of formation(HOFs) of the reference compoundsa
表2列出3,3′-偶氮-1,2,4,5-四嗪衍生物计算的总能量、零点能、温度校正项以及生成热.结果显示,B3P86/ 6-311G**和B3LYP/6-311G**这两种方法计算的生成热结果十分接近,具有很好的线性关系:HOFB3P86= 1.0003HOFB3LYP-6.7479,R2=0.9997.这说明两种方法对标题化合物生成热的计算结果都较为理想.两种方法对于B1(DAAT)的HOF计算结果与实验值[11]都比较接近,前者略大于后者约50 kJ·mol-1且在误差范围内(±48 kJ·mol-1),见表2.同时与先前报道的B1理论计算结果1148.51 kJ·mol-1[8]相比,我们计算的结果更加接近实验值.这也表明了两种方法用以预测标题化合物的生成热数据是可靠的.从表2可见,所有的3,3′-偶氮-1,2,4,5-四嗪衍生物都具有高正生成热,超过1000 kJ·mol-1,其中B4具有最高的生成热大约1830 kJ·mol-1.符合先前的研究报道,含能高氮杂环化合物具有高正生成热[1-2].标题化合物的生成热数值远高于我们之前研究的1,2,4,5-四嗪衍生物[4],原因可能为前者分子结构中含有偶氮基以及两个四嗪环.—NHNH2、—NO2、—N3或—NHNO2取代的衍生物生成热比未取代的3,3′-偶氮-1,2,4,5-四嗪生成热高,而—NH2或—NF2取代的衍生物生成热相对较低.在带有相同取代基数量的情况下,带有—N3取代基的衍生物生成热最大,这与先前的研究结果一致:—N3基团可以显著增加分子的生成热,是已知含能材料官能团中增加生成热显著的一类取代基,大约增加293-320 kJ·mol-1[4-5,9,36].通过对比单取代和双取代衍生物的生成热,我们发现3,3′-偶氮-1,2,4,5-四嗪衍生物的生成热并不符合简单的基团加和规律.
2.2 热稳定性
键级是基于分子轨道系数进行Mulliken布居数分析所得的重要电子结构参数.一般而言,原子间键级越大,表明其间电子云密度较大或重叠较多,预示该键较强、不易断裂.表3给出标题化合物分子的C—N(四嗪环)、N—N(四嗪环)、N7=N8、C—R(R=—NH2、—NHNH2、—NO2、—N3、—NHNO2、—NF2)以及N—R′(R′=—NH2、—N2、—NO2)的重叠布居数.结果显示,C—R或N—R′键的键级大都比C—N、N—N和N7=N8键的键级小(衍生物带有N—R′键时不比较C—R键),只有A1、B1和B2的C—R或N—R′键的键级比N7=N8键级大.A3、A6、B3、B6以及C1—C5的C—R键级在所有化学键中最小,表明在这些衍生物中C—R键较弱,在热解或撞击下可能最易断裂,是热解引发键.A2、A4、A5、B4和B5的N—R′键具有最小的布居数,因此热解时可能生成R′自由基.然而,A1、B1和B2的N7=N8键级最小,表明热解时连接两个四嗪环的偶氮键可能作为引发键发生断裂.我们发现,大多数衍生物N—N键的键级比3,3′-偶氮-1,2,4,5-四嗪大,除了A3、A6、B3、B4、B6、C3和C5,说明取代基的引入可以增加3,3′-偶氮-1,2,4,5-四嗪N—N键的强度.在所有标题化合物中,A1最弱键的键级(0.2615)最大.根据判别撞击感度相对大小的“最小键级原理(PSBO)”[19],预示A1的感度最小、热力学稳定性最好.

表2 计算的3,3′-偶氮-1,2,4,5-四嗪衍生物计算的总能量(E0)、零点能(ZPE)、温度校正项(HT)和生成热(HOFs)aTable 2 Calculated total energies(E0,),zero-point energies(ZPE),thermal corrections(HT),and heats of formation(HOFs) of the 3,3′-azobis-1,2,4,5-tetrazine derivatives at the B3LYPand B3P86 levelsa

表3 3,3′-偶氮-1,2,4,5-四嗪衍生物在B3LYP/6-311G**水平上计算的部分键平均重叠布居数(P)Table 3 Calculated average bond overlap populations(P) of part bonds of the 3,3′-azobis-1,2,4,5-tetrazine derivatives at the B3LYP/6-311G**level
选择Mulliken布居数最小的C—R、N—N或N—R′键作为可能的热解引发键,用UB3LYP/6-311G**方法计算这些键断裂所需的离解能(BDE),标题化合物最弱键的重叠布居数以及键离解能(BDE)列于表4.结果表明没有零点能校正的键离解能(BDE0)通常比校正的键离解能(BDEZPE)大,但零点能不影响化合物的稳定性次序.当3,3′-偶氮-1,2,4,5-四嗪被—NH2和—N3基团取代后BDEZPE有所增加,而其它衍生物的BDEZPE比未取代的3,3′-偶氮-1,2,4, 5-四嗪小.计算的BDE值可以用来比较含能材料的相对热稳定性顺序[37-38].因此,可以推断出—NH2和—N3基团有利于增加3,3′-偶氮-1,2,4,5-四嗪的热稳定性.通过与常用炸药RDX(黑索今,环三亚甲基三硝胺)以及HMX(奥克托今,1,3,5,7-四硝基-1,3,5,7-四氮杂环辛烷)比较,我们发现大多数衍生物具有较高的BDEZPE,这表明它们具有较为理想的热稳定性.
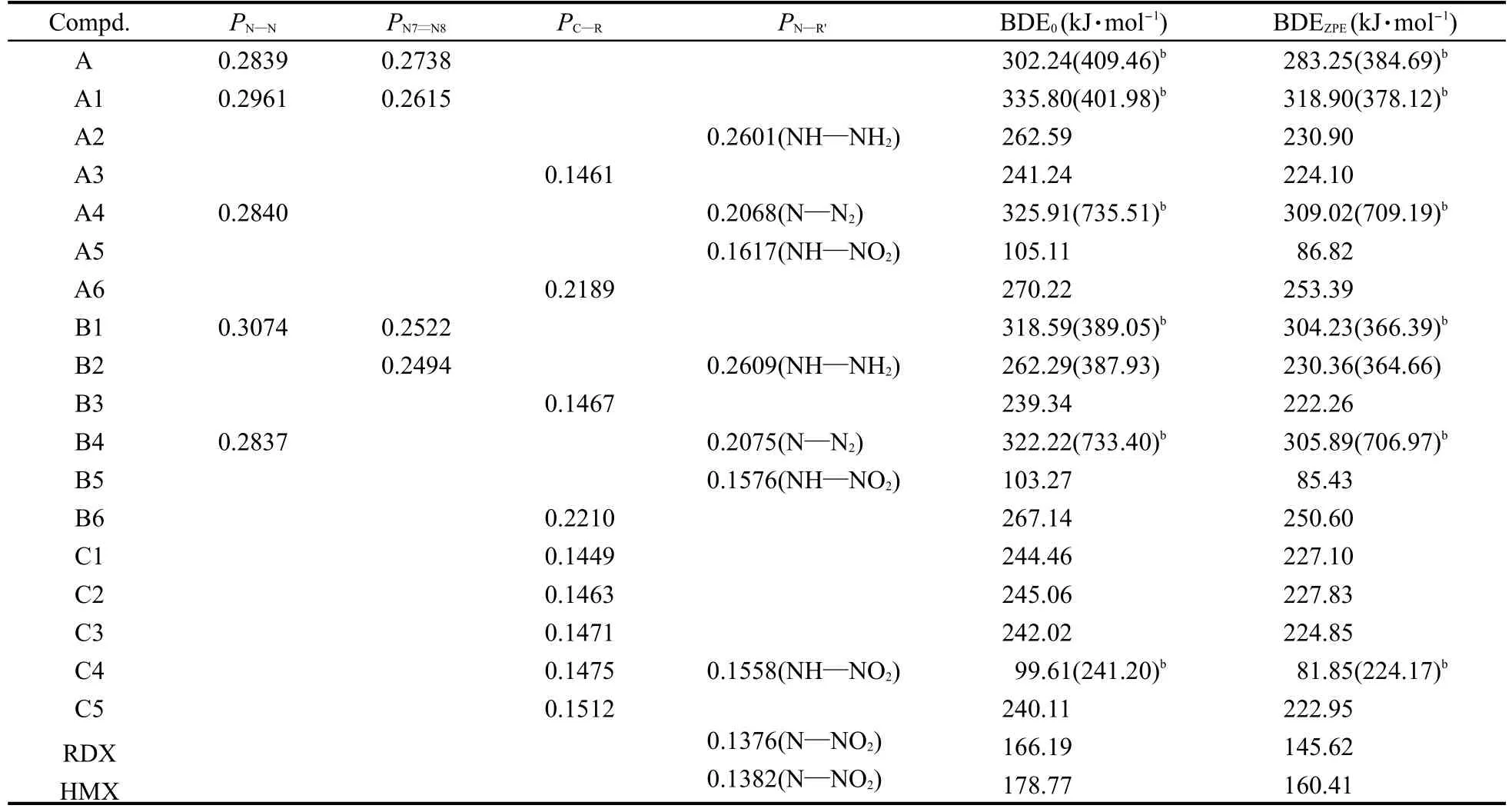
表4 在B3LYP/6-311G**水平上计算的3,3′-偶氮-1,2,4,5-四嗪衍生物以及RDX和HMX的最弱键重叠布居数以及键离解能(BDE)aTable 4 Calculated bond dissociation energies(BDE)and bond overlap populations of the weakest bonds for the 3,3′-azobis-1,2,4,5-tetrazine derivatives together with RDX and HMX at the B3LYP/6-311G**levela
分析结果发现A4的N—R′具有相对较低的键级(0.2068),但在所有取代3,3′-偶氮-1,2,4,5-四嗪衍生物的最弱键中,A4具有最大的BDEZPE值(709.19 kJ·mol-1).然而,虽然A4四嗪环上N—N键具有较高的键级(0.2840),但却具有相对较低的BDEZPE值(309.02 kJ·mol-1).B4的计算结果与A4相似,表明A4和B4在受热或撞击下应该发生四嗪环断裂.由此说明,在判定标题化合物热稳定性时不能只是简单依据化学键的布居数,有必要借助键离解能来判定.从表4可以看出除了A1和B1,B系列衍生物的BDE与A系列衍生物的BDE非常接近,说明增加取代基数量对衍生物的稳定性影响较小.同时与之前研究的单环1,2,4,5-四嗪衍生物结果[4]相比较,我们发现相同取代基对于不同体系稳定性的影响有所不同,在单环1,2,4,5-四嗪衍生物中其稳定性随着取代基数目的增加而增加.另外,B1四嗪环上N—N的键级(0.3074)比N7=N8的键级(0.2522)高,但前者的BDE比后者低,说明B1热解时首先发生四嗪环断裂,与先前的报道相符[11].同时说明在键离解能数据没有相关实验值的情况下,其数值与传统炸药在相同计算水平UB3LYP/6-311G**的所得数据相比,可用以判定新设计的偶氮四嗪衍生物的热稳定性是否理想,先前的工作已证明该计算方法是可行的[4-5].
2.3 爆轰性能
含能材料的爆速和爆压是两个重要的性能参数,预测的爆速(D)和爆压(p)列于表5.为了进行比较,已知爆轰性能实验值的RDX和HMX也列于表5[39].先前的研究表明[4,5,4042],将半经验的K-J方程应用于预测高氮化合物的爆轰性能是可靠的.

表5 3,3′-偶氮-1,2,4,5-四嗪衍生物以及RDX和HMX的爆轰性能aTable 5 Predicted detonation properties of the 3,3′-azobis-1,2,4,5-tetrazine derivatives together with RDX and HMXa
如表5所示,B1、RDX以及HMX的理论预测值(ρ、D和p)与实验值[12,39]比较接近,说明该方法用于预测标题化合物的爆轰性能是相对可靠的.结果显示,带有不同取代基团的3,3′-偶氮-1,2,4,5-四嗪衍生物其密度不同,最大的ρ为1.95 g·cm-3,最小的ρ为1.61 g·cm-3,这也同时说明了标题化合物可能具有不同的D和p.除了A1、A2和B1,其他衍生物的爆速和爆压高于未取代的3,3′-偶氮-1,2,4,5-四嗪.通过比较A系列与B、C系列衍生物的数据,我们发现当增加取代基数目时ρ、D和p有所增加,除了带有—NH2取代基的衍生物以及C2.从表5可以看出B3、B6、B7和C4-C6的密度比RDX(1.82 g·cm-3)高,所以它们应当具有较高的D和p.带有—NO2或—NF2基团的衍生物表现出较高的ρ、D以及p,其值分别接近于1.90 g·cm-3、9.00 km·s-1以及40.00 GPa.在所有的衍生物中带有两个—NF2基团的B6具有最大的ρ、D以及p.这表明—NO2或—NF2取代基有利于增加3,3′-偶氮-1,2,4,5-四嗪衍生物的密度以及爆轰性能.
2.4 预测潜在的HEDM候选物
与传统的含能材料(含碳的氧化物)相比,大多数高氮化合物都具有非常高的正生成热[43].然而,高生成热不利于化合物的稳定性.因此,一种理想的富氮HEDM候选物不仅要具有优良的爆轰性能而且还应具备较好的稳定性.图2给出了3,3′-偶氮-1,2,4, 5-四嗪衍生物的爆轰性能以及最弱键离解能,也包括RDX和HMX的数据.
从图2可以看出,B3、B6、C4和C5的D和p比 RDX高,仅B6有非常理想的爆轰性能,它的D和p超过HMX.与RDX和HMX相比,大多数3,3′-偶氮-1,2,4,5-四嗪衍生物具有较高的最弱键离解能.基于热解引发键的BDE数据,可以判定大多数标题化合物在热解和撞击下可以表现出较低的感度,即稳定性较好.综上所述,可以得出结论:B3、B6和C5具有良好的爆轰性能(D和p)以及热稳定性(BDE)接近于RDX和HMX.因此,B3、B6和C5可以作为潜在低感度以及高爆轰性能的品优HEDM候选物.
3 结论
通过使用B3LYP/6-311G**和B3P86/6-311G**方法计算研究了系列3,3′-偶氮-1,2,4,5-四嗪衍生物的生成热.结果表明:两种方法得到的结果十分接近,—N3取代基可显著增加3,3′-偶氮-1,2,4,5-四嗪衍生物的生成热.通过分析标题化合物的最弱键离解能,我们发现大多数标题化合物具有较为理想的热稳定性,其中—NH2和—N3取代基非常有利于增加衍生物的热稳定性,但是3,3′-偶氮-1,2,4,5-四嗪被—NHNO2取代后情况却相反.比较爆速和爆压计算结果显示:—NO2或—NF2取代基有利于增加3,3′-偶氮-1,2,4,5-四嗪衍生物的爆轰性能.综合爆轰性能和热稳定性的结果,B3、B6和C5可以作为潜在的品优HEDM候选物.以上结果对设计合成品优HEDM及其性能研究具有参考价值和指导意义.
1 Huynh,M.H.V.;Hiskey,M.A.;Pollard,C.J.;Montoya,D.P.; Hartline,E.L.;Gilardi,R.D.J.Energ.Mater.,2004,22:217
2 Huynh,M.H.V.;Hiskey,M.A.;Archuleta,J.G.;Roemer,E.L.; Gilardi,R.D.Angew.Chem.Int.Edit.,2004,43:5658
3 Talawar,M.B.;Sivabalan,R.;Senthilkumar,N.;Prabhu,G.; Asthana,S.N.J.Hazard.Mater.,2004,113:11
4 Wei,T.;Zhu,W.H.;Zhang,X.W.;Li,Y.F.;Xiao,H.M.J.Phys. Chem.A,2009,113:9404
5 Wei,T.;Zhu,W.H.;Zhang,J.J.;Xiao,H.M.J.Hazard.Mater., 2010,179:581
6 Chavez,D.E.;Hiskey,M.A.;Gilardi,R.D.Org.Lett.,2004,6: 2889
7 Chavez,D.E.;Hiskey,M.A.J.Energ.Mater.,1999,17:357
8 Wilcox,C.F.;Zhang,Y.X.;Bauer,S.H.J.Energ.Mater.,2002, 20:71
9 Chavez,D.E.;Hiskey,M.A.;Gilardi,R.D.Angew.Chem.Int. Edit.,2000,39:1791
10 Kerth,J.;Löbbecke,S.Propellants Explos.Pyrotech.,2002,27: 111
11 Löbbecke,S.;Schuppler,H.;Schweikert,W.J.Therm.Anal. Calorim.,2003,72:453
12 Chavez,D.E.;Hiskey,M.A.;Naud,D.L.Propellants Explos. Pyrotech.,2004,29:209
13 Rice,B.M.;Hare,J.Thermochim.Acta,2002,384:377
14 Muthurajan,H.;Sivabalan,R.;Talawar,M.B.;Anniyappan,M.; Venugopalan,S.J.Hazard.Mater.,2006,133:30
15 Hohenberg,P.;Kohn,W.Phys.Rev.B,1964,136:864
16 Kohn,W.;Sham,L.J.Phys.Rev.A,1965,140:1133
17 Salahub D.R.;Zerner,M.C.The challenge of d and f electrons. Washington D.C.:ACS,1989
18 Parr,R.G.;Yang,W.Density-functional theory of atoms and molecules.Oxford:Oxford University Press,1989:1-333
19 Chen,Z.X.;Xiao,J.M.;Xiao,H.M.;Chiu,Y.N.J.Phys.Chem. A,1999,103:8062
20 Xiao,H.M.;Chen,Z.X.The modern theory for tetrazole chemistry.Beijing:Science Press,2000:128-158 [肖鹤鸣,陈兆旭.四唑化学的现代理论.北京:科学出版社,2000: 128-158]
21 Chen,P.C.;Chieh,Y.C.;Tzeng,S.C.J.Mol.Struct.-Theochem, 2003,634:215
22 Ju,X.H.;Li,Y.M.;Xiao,H.M.J.Phys.Chem.A,2005,109:934 23 Hahre,W.J.;Radom,L.;Schleyer,P.V.R.;Pole,J.A.Ab initio molecular orbital theory.New York:Wiley-Interscience,1986
24 Wang,F.;Xu,X.J.;Xiao,H.M.;Zhang,J.Acta Chim.Sin., 2003,61:1939 [王 飞,许晓娟,肖鹤鸣,张 骥.化学学报, 2003,61:1939]
25 Ju,X.H.;Wang,X.;Bei,F.L.J.Comput.Chem.,2005,26:1263
26 (a)David,R.L.Handbook of chemistry and physics.84th ed. Gaithersburg,MD:CRC Press,2003-2004:sect 5 (b)Afeefy,H.Y.;Liebman,J.F.;Stein,S.E.“Neutral thermochemical data”in NIST chemistry webbook,NIST standard reference database number 69.Linstrom,P.J.;Mallard,W.G.Eds. Gaithersburg,MD:National Institute of Standards and Technology,2000(http://webbook.nist.gov) (c)Lias,S.G.;Bartmess,J.E.;Liebman,J.F.;Holmes,J.L.; Levin,R.D.;Mallard,W.G.J.Phys.Chem.Ref.Data,1988: Suppl.No.1
27 Curtiss,L.A.;Raghavachari,K.;Trucks,G.W.;Pople,J.A. J.Chem.Phys.,1991,94:7221
28 Curtiss,L.A.;Raghavachari,K.;Redfern,P.C.;Pople,J.A. J.Chem.Phys.,1997,106:1063
29 Benson,S.W.Thermochemical kinetics.2nd ed.New York: Wiley-Interscience,1976
30 Mills,I.;Cvitas,T.;Homann,K.;Kallay,N.;Kuchitsu,K. Quantities,units,and symbols in physical chemistry.Oxford: Blackwell Scientific Publications,1988:1-233
31 Blanksby,S.J.;Ellison,G.B.Acc.Chem.Res.,2003,36:255
32 Kamlet,M.J.;Jacobs,S.J.J.Chem.Phys.,1968,48:23
33 Rice,B.M.;Hare,J.J.;Byrd,E.F.C.J.Phys.Chem.A,2007, 111:10874
34 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09. RevisionA.01.Wallingford,CT:Gaussian Inc.,2009
35 Scott,A.P.;Radom,L.J.Phys.Chem.,1996,100:16502
36 Huynh,M.H.V.;Hiskey,M.A.;Chavez,D.E.;Naud,D.L.; Gilardi,R.D.J.Am.Chem.Soc.,2005,127:12537
37 Owens,F.J.J.Mol.Struct.-Theochem,1996,370:11
38 Rice,B.M.;Sahu,S.;Owens,F.J.J.Mol.Struct.-Theochem, 2002,583:69
39 Talawar,M.B.;Sivabalan,R.;Mukundan,T.;Muthurajan,H.; Sikder,A.K.;Gandhe,B.R.;Subhananda,R.A.J.Hazard. Mater.,2009,161:589
40 Türker,L.;Atalar,T.;Gümüş,S.;Çamur,Y.J.Hazard.Mater., 2009,167:440
41 Smith,M.W.;Cliff,M.D.NTO-Based explosive formulations:a technology review.Salisbury South,Australia:DSTO-TR-0796, 1999:19-20
42 Gálvez-Ruiz,J.C.;Holl,G.;Karaghiosoff,K.;Klapötke,T.M. Löhnwitz,K.;Mayer,P.;Nöth,H.;Polborn,K.;Rohbogner,C.J.; Suter,M.;Weigand,J.J.Inorg.Chem.,2005,44:4237
43 Zhang,M.X.;Eaton,P.E.;Gilardi,R.D.Angew.Chem.Int. Edit.,2000,39:401
July 19,2010;Revised:September 19,2010;Published on Web:November 1,2010.
Molecular Design of 3,3′-Azobis-1,2,4,5-tetrazine-Based High-Energy Density Materials
ZHANG Jing-Jing1GAO Hong-Wei2WEI Tao2,*WANG Chao-Jie2
(1Renji College,Wenzhou Medical College,Wenzhou 325035,Zhejiang Province,P.R.China;2Department of Pharmacy, Wenzhou Medical College,Wenzhou 325035,Zhejiang Province,P.R.China)
We systematically studied the heats of formation(HOFs)for a series of 3,3′-azobis-1,2,4, 5-tetrazine derivatives by density functional theory(DFT).The results show that the—N3group plays a very important role in increasing the HOFs for these derivatives.An analysis of the bond dissociation energies for the weakest bonds indicates that the attachment of—NH2or—N3group to 3,3′-azobis-1,2,4, 5-tetrazine is favorable in enhancing its thermal stability.The calculated detonation velocities(D)and pressures(p)indicates that—NO2or—NF2largely enhances the detonation performance of the derivatives.Considering the detonation performance and the thermal stability,the three derivatives may be regarded to be promising candidates for high-energy density materials(HEDMs).
Density functional theory;3,3′-Azobis-1,2,4,5-tetrazine;Heat of formation; Bond dissociation energy;Detonation property
O641
∗Corresponding author.Email:weitao@wzmc.edu.cn;Tel:+86-577-86689984.
The project was supported by the Natural Science Foundation of Zhejiang Province,China(Y5080043).
浙江省自然科学基金(Y5080043)资助项目
猜你喜欢
高中数理化(2022年2期)2022-02-22
世界农药(2019年3期)2019-09-10
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
中国塑料(2016年11期)2016-04-16
中国塑料(2016年7期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
应用化工(2014年1期)2014-08-16
无机化学学报(2014年6期)2014-02-28
