
金属离子(Na+、K+、Ca2+、Mg2+、Zn2+)与鸟嘌呤异构体配合物的稳定性
2010-11-30 10:49:10赵永平艾洪奇陈金鹏杨爱彬齐中囡
物理化学学报 2010年12期
赵永平 艾洪奇 陈金鹏 杨爱彬 齐中囡
(济南大学化学化工学院,济南 250022)
金属离子(Na+、K+、Ca2+、Mg2+、Zn2+)与鸟嘌呤异构体配合物的稳定性
赵永平 艾洪奇*陈金鹏 杨爱彬 齐中囡
(济南大学化学化工学院,济南 250022)
在B3LYP/6-311++G**水平上用极化连续介质模型(PCM)系统研究了金属离子(M+/2+=Na+,K+,Ca2+, Mg2+,Zn2+)和十三种鸟嘌呤异构体形成的配合物GnxM+/2+(n为鸟嘌呤异构体的编号,x表示M+/2+与鸟嘌呤异构体的结合位点)在气(g)液(a)两相中的稳定性顺序.着重探讨了液相中配合物的稳定性差异,并且从溶质-溶剂效应、结合能、形变能及异构体的相对能量等几个方面分析了造成稳定顺序发生变化的原因.报道了溶液中这五种金属离子与鸟嘌呤异构体结合形成的六种基态配合物:aG1N2,N3Na+,aG1N2,N3K+,aG1O6,N7Ca2+,aG1N2,N3Mg2+(aG1O6,N7Mg2+),aG2N3,N9Zn2+.可以看出,除了在Zn2+配合物中鸟嘌呤异构体为G2外,构成其余四种金属离子配合物的鸟嘌呤异构体都是G1,但结合位点不同.同时对气相中各类配合物稳定性也进行了系统的排序,并报道了几种较稳定的配合物,如:gG3N1,O6K+,gG5N1,O6K+,gG3N1,O6Ca2+/Mg2+,gG4O6,N7Ca2+/Mg2+.
鸟嘌呤异构体;稳定性;金属离子;溶质-溶剂效应;结合能
金属离子-碱基相互作用参与了许多的生理过程,如维持三倍体、四倍体DNA螺旋的稳定性以及Z-DNA的稳定性[1];一些金属离子可以扰乱DNA的复制过程[2],这与金属离子和DNA碱基对相互作用的稳定性有关[3-4].碱金属及碱土金属离子能够引发碱基上电子的重新分布,使部分高能异构体趋于稳定[5];这些高能异构体的存在增加了DNA碱基错配的可能性,导致一些自发的基因突变[7].Pettitt等[7]研究GC碱基对时发现鸟嘌呤N7位上结合二价金属离子时其N1位上的氢会转移到胞嘧啶的N3位. Oliva等[8]发现三水合金属离子的结合可以明显地提高气相中GC对和G碱基的稳定性.
Sabio等[9]对各类鸟嘌呤异构体的模拟计算发现,烯酮式鸟嘌呤异构体的能量要比最稳定的烯醇式异构体的能量至少低8.4 kJ·mol-1,这和Choi等[10]用红外线激光器的研究结果一致.而Hanus等[11]的研究发现,在气相和一水或二水条件下,鸟嘌呤各异构体的稳定顺序基本不变,但在溶剂条件下,三种稀有异构体的稳定性要大于典型鸟嘌呤异构体.
Kabelac等[12]在研究Na+、Mg2+、Zn2+与各鸟嘌呤异构体相互作用时发现,气相中典型鸟嘌呤异构体的金属离子配合物的稳定能比稀有鸟嘌呤异构体的要高.Mazzuca等[13]研究发现,金属离子Al3+和核酸碱基异构体相互作用导致鸟嘌呤的高能异构体变得相当稳定.Pedersen等[14]采用两类稀有鸟嘌呤异构体与Al3+相互作用时,发现烯酮式鸟嘌呤异构体复合物比烯醇式的要稳定.Russo等在研究气相中一价金属离子M+=Na+/K+与五种鸟嘌呤异构体的相互作用时发现所得配合物的稳定性顺序为G1M+>G2M+>G7M+>G6M+>G8M+,这与鸟嘌呤异构体的自身的顺序G2>G1>G6>G8>G7有很大不同[15];二价金属离子Ca2+和Mg2+配合物的稳定性顺序[16]为G1M2+>G7M2+>G2M2+>G6M2+>G8M2+,也表现了很大不同.
由此可见,人们对鸟嘌呤异构体金属离子配合物的稳定性研究主要集中在气相或微水环境[17-18](体系中含有一到五个水分子).微水环境只是局部的水合作用,与整个水合的环境还有很大的区别[19].鸟嘌呤异构体在微水环境和整个水合环境稳定性表现出很大的差异就已证明了这一点[11].考虑到整个水溶剂环境中配合物稳定性研究目前尚未见报道,本文将研究M+/2+=Na+,K+,Ca2+,Mg2+,Zn2+五种金属离子与各鸟嘌呤异构体形成的配合物在整个水溶液中的稳定性顺序以及其影响因素,找出溶液中这些配合物的最稳定存在形式,并与气相中的稳定性结果进行了比较.
1 计算模型和方法
计算是用Gaussian 03程序[20]在联想Pentium IV计算机上采用B3LYP/6-311++G**//6-31+G*方法进行.采用PCM模型模拟整个水溶液环境.Hunter等[19]和Kumar等[21]曾利用这一方法和模型开展过对GC碱基对溶剂效应的研究,艾洪奇等[22]曾利用此方法研究了在溶液中锌离子与腺嘌呤异构体配合物的稳定性顺序,Gustavsson等[23]以水为溶剂利用PCM模型优化了十一种尿嘧啶异构体,计算所得异构体激发态寿命结果与在紫外荧光检测仪测得的数据非常一致,从而证实了PCM模型在该类体系中的可靠性.
Shukla等[24]用PCM模型计算了四种(aG1,aG2, aG6,aG7)鸟嘌呤异构体的相对能量,Hanus等[11]则用COSMORS模型计算了七种(aG1-aG7)异构体的相对能量,发现两实验中所得aG1,aG2,aG6,aG7四种异构体的稳定顺序一致,为了验证所选模型对所研究体系的可用性,本文又选用了PCM模型结合六种原子半径(UAO,UAHF,UFF,PAULING,BONDI, KLAMT)和COSMORS模型默认原子半径(KLAMT)模拟计算了aGnxNa+前六种配合物的相对能量.由Supporting Information(SI)表S1中各模型的相对能量值可以看出,除了采用BONDI半径的PCM模型的第二三位配合物的稳定顺序发生颠倒,其余五种原子半径的PCM模型计算所得配合物稳定顺序一致.Gustavsson等[23,25]在研究尿嘧啶时也已经证明了原子半径的选择并不影响实验结果,采用KLAMT半径的COSMORS模型计算所得配合物的稳定顺序与采用UAO半径PCM模型所得顺序也非常一致.
因此为了便于与已知的液相中鸟嘌呤异构体的实验数据[11,24]进行比较,本文继续采用了UAO原子半径及PCM模型,以水作为溶剂(ε=78.39,ε为介电常数)计算了十三种鸟嘌呤异构体及其各种金属配合物,其中鸟嘌呤异构体选取是根据文献[10-12, 15]所得.体现稳定性的相对能量选择了经零点能修正后的能量.
考虑到溶液中配合物的稳定性主要由溶质-溶剂作用能量值(Es)、结合能(Eb)、形变能(Ed)及鸟嘌呤异构体自由能(EG)这四项决定,故拟用这四项能量值的相对大小来分析构成配合物稳定性变化的主要原因,即ΔEbsd=ΔEs+ΔEb+ΔEd+ΔEG.加上“Δ”表示取相对值,如ΔEbsd为考虑上述四种因素后得到的相对能量.Eb为经BSSE校正后配合物能量EGnM+/2+(BSSE很小,用气相中得到的BSSE校正)与各反应物能量和之差[26],即定义为配合物中鸟嘌呤的能量(EGn')与其相应自由鸟嘌呤的能量之差,其中n表示鸟嘌呤异构体的编号,n=1,2,…, 13.
图1显示了得到的液气两相中十三种鸟嘌呤异构体与五种金属离子(M+/2+=Na+,K+,Ca2+,Mg2+,Zn2+)相互作用得到的290种配合物GnxM+/2+,其中x表示M+/2+与鸟嘌呤异构体的结合位点.每一种异构体都根据金属离子结合位点的不同可得到二到三种金属配合物.后面正文中只对每一aGn中最稳定的配合物进行了稳定性排序及原因分析.如G1异构体可在其N2,N3及O6,N7处分别结合一个金属离子形成G1N2,N3M+/2+和G1O6,N7M+/2+两种配合物(详见图1中第一行左侧),原子的编号详见图中第一个配合物.为叙述方便,气(g)液(a)两相中的配合物分别加上了这两个前缀,如gGnxM+/2+和aGnxM+/2+.
2 结果与讨论
2.1 液相中的稳定性排序及影响因素
在SI表S2中列出了液气两相中G异构体及GnxM+(M+=Na+,K+)配合物的各项能量.结果显示:液相中各鸟嘌呤异构体的稳定性顺序为aG1>aG2>aG3>aG4>aG6>aG7>aG8>aG5>aG12>aG9>aG10>aG11>aG13.可见,Hanus等[11]用COSMORS模型计算的aG1,aG2,aG3,aG4,aG5,aG6,aG7稳定顺序,及Shukla等[24]用B3LYP方法和PCM模型计算的四种鸟嘌呤异构体的相对自由能aG1(0.0 kJ·mol-1), aG2(2.2 kJ·mol-1),aG6(29.3 kJ·mol-1),aG7(35.3 kJ· mol-1)都与我们的计算值相近,且稳定顺序完全一致(详见表S2).从而验证了该模型在GnxM+/2+体系中的适用性.
结合Na+后配合物稳定性顺序变为aG1N2,N3Na+>aG2O6Na+>aG3N1,O6Na+>aG4O6,N7Na+>aG5N1,O6Na+>aG6O6,N7Na+>aG7N3,N9Na+>aG8N1,O6Na+>aG9O6,N7Na+>aG10N1,O6Na+>aG11N1,O6Na+>aG12N2,N3Na+>aG13O6,N7Na+.为讨论方便,本文仅以最稳定的前六种配合物为例(下同),将它们液相中以aG1N2,N3M+为基准得到的各项相对能量指标列于表S3,取其前六种最稳定配合物的各相对能量列于表1.比较显示由Na+和K+配合物的ΔEbsd所得稳定顺序与由其相应的配合物自由能相对值(ΔE)得到的顺序非常一致,说明用ΔEbsd的构成来分析配合物的稳定是合理的.aG1N2,N3Na+的ΔEs和ΔEb比aG2O6Na+的分别要低10.8、3.7 kJ·mol-1,且aG1的ΔEG比aG2的低3.4 kJ·mol-1,由此可见,是ΔEs,ΔEb和ΔEG共同决定了aG1N2,N3Na+的稳定性大于aG2O6Na+.aG3N1,O6Na+的ΔEs和ΔEb比aG4O6,N7Na+的分别高1.2、3.4 kJ·mol-1,而aG3的ΔEG比aG4的低13.8 kJ·mol-1,可见是ΔEG抵消了ΔEs和ΔEb的不利影响并决定了aG3N1,O6Na+的稳定性大于aG4O6,N7Na+.由于aG5的自由能比aG6高8.6 kJ·mol-1,其配合物的ΔEs和ΔEb比aG6O6,N7Na+分别低2.1和10.4 kJ·mol-1,因此ΔEs和ΔEb抵消了ΔEG的升高,并决定了aG5N1,O6Na+比aG6O6,N7Na+稳定.总的来说,形变能贡献都不大.由此可见,这几个影响因素在各个配合物的稳定性排序上面发挥的作用各不相同.
与aGnxNa+配合物顺序相比,aGnxK+配合物中只有aG5N1,O6K+与aG6O6,N7K+的顺序发生颠倒.分析发现,aG5N1,O6K+的稳定性降低的原因是其ΔEs和ΔEG比aG6O6,N7K+的分别高2.5和8.6 kJ·mol-1,故不利于aG5N1,O6K+的稳定,尽管该配合物中K+与G5的结合强度比与G6的强5.1 kJ·mol-1,但仍不足以补偿ΔEs和其异构体ΔEG引起的配合物能量升高.
SI中表S4列出了液气两相中GnxM2+配合物(M=Mg,Ca,Zn)的各项能量.其中aGnxCa2+配合物的稳定性顺序为aG1O6,N7Ca2+>aG2N3,N9Ca2+>aG4O6,N7Ca2+>aG5N1,O6Ca2+>aG3N1,O6Ca2+>aG7N3,N9Ca2+>aG9O6,N7Ca2+>aG10N1,N2Ca2+>aG12N7Ca2+>aG6N1,N2Ca2+>aG8N2,N3Ca2+>aG13O6,N7Ca2+>aG11N7Ca2+.与aGnxNa+配合物稳定性顺序相比较,排在前五位的只有aG3N1,O6Ca2+发生了变化,排到了第五位,而排在第六至十三位的配合物稳定性顺序有较大的变化.表2列出了液相中以aG1O6,N7M2+为基准得到的前六个最稳定配合物的各项相对能量指标(全部十三种aG1xM2+的各相对能量详见表S5中),ΔEG值与表1中相同.比较显示, aG2N3,N9Ca2+比aG1O6,N7Ca2+能量高的原因是其不利的ΔEs(16.9 kJ·mol-1),ΔEb(15.5 kJ·mol-1)和ΔEG(3.4 kJ· mol-1)能量,而其低的ΔEd(-23.8 kJ·mol-1)不足以补偿这些损失所致.aG3N1,O6Ca2+变得不如aG5N1,O6Ca2+稳定的原因是,后者在溶液中的ΔEs(9.6 kJ·mol-1)和ΔEb(-13.0 kJ·mol-1)分别比aG3N1,O6Ca2+降低了18.1、30.6 kJ·mol-1,弥补了其不利的ΔEd(0.8 kJ·mol-1)和ΔEG5(39.7 kJ·mol-1)贡献,从而致使二者稳定顺序发生变化.aG4O6,N7Ca2+和aG5N1,O6Ca2+的稳定性排在第四和第五位置的原因与一价的aG4O6,N7Na+和aG5N1,O6Na+类似,不再赘述.aG6N1,N2Ca2+的稳定性跌至第九主要是由其异常高的形变能(ΔEd=54.0 kJ·mol-1)和不稳定的异构体能(ΔEG=31.1 kJ·mol-1)造成的.
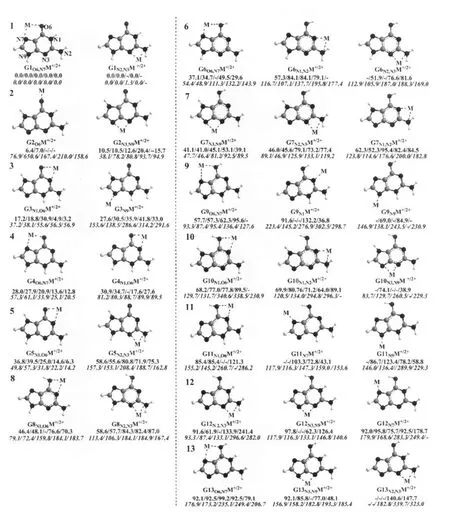
图1 GnxM+/2+配合物优化构型及其相对自由能Fig.1 Geometries and relative free energies of GnxM+/2+complexes M denotes the metal ion,and active sites of guanine isomers bound by the metal ions are associated with dashed lines.Those complexes formed by identical guanine isomer with its different active sites are put on the same row,thus 13 rows of complexes formed by 13 guanine isomers are displayed according to their relative energy orderings.Relative energies in aqueous(in bold)and gas phases(in bold and italic)are listed under each corresponding complex and shown as x1/x2/x3/x4/x5 forms,which denote the relative energies of Na+/K+/Ca2+/Mg2+/Zn2+-complexes, respectively.“-”indicates that the complex is absent or not obtained.Energy is in kJ·mol-1.
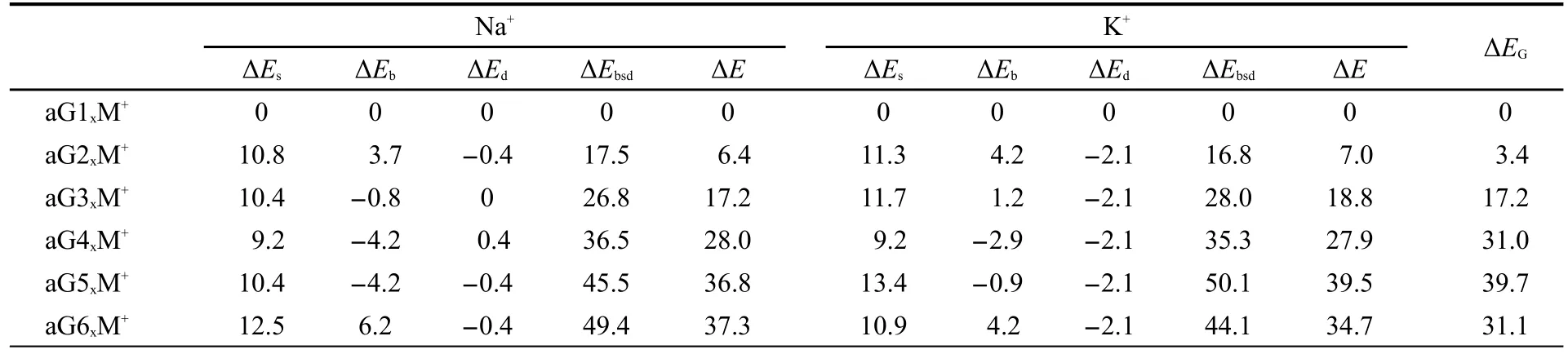
表1 液相中配合物aGnxM+的各项相对能量Table 1 Various relative energies of complexes aGnxM+
在液相中aGnxMg2+配合物的稳定性的顺序为aG1N2,N3Mg2+=aG1O6,N7Mg2+>aG3N1,O6Mg2+>aG4O6,N7Mg2+>aG5N1,O6Mg2+>aG2N3,N9Mg2+>aG6O6,N7Mg2+>aG7N3,N9Mg2+>aG12N3,N9Mg2+>aG10N2,N3Mg2+>aG11N7Mg2+>aG8N1,O6Mg2+>aG13N3,N9Mg2+>aG9N9Mg2+.即溶液中Mg2+与G1的N2, N3或O6,N7位双齿结合得到的配合物稳定性几乎是相同的,而Ca2+与G1结合时只得到aG1O6,N7Ca2+.由表2数据表明,aG2N3,N9Ca2+的稳定性在其配合物中排第二,而aG2N3,N9Mg2+则下降到了第五.原因是:排到第二至第四的配合物aG3N1,O6Mg2+、aG4O6,N7Mg2+、aG5N1,O6Mg2+的ΔEs和ΔEb都远低于aG2N3,N9Mg2+的相应值所致.aG6O6,N7Mg2+的稳定性依然排在第六位是其弱的ΔEb和高的ΔEG所致,该原因与aG2N3,N9Mg2+在其异构体稳定性中排在第五位的原因有所不同
在液相中aGnxZn2+配合物的稳定性的顺序为aG2N3,N9Zn2+>aG1O6,N7Zn2+>aG3N1,O6Zn2+>aG5N1,O6Zn2+>aG4O6,N7Zn2+>aG6O6,N7Zn2+>aG9N1Zn2+>aG10N3,N9Zn2+>aG7N3,N9Zn2+>aG11N7Zn2+>aG13N3,N9Zn2+>aG8N1,O6Zn2+>aG12N3,N9Zn2+,其中最稳定的构型aG2N3,N9Zn2+对应的鸟嘌呤异构体是aG2,而与前面四种金属离子形成最稳定配合物的异构体是aG1.aG1在液相中的稳定性排第一[11,24].表2显示,aG2N3,N9Zn2+配合物的各项能量指标(ΔEs,ΔEb,ΔEd和ΔEG)都比aG1O6,N7Zn2+配合物的低,说明G2构型非常适合Zn2+的结合,从而使得aG2N3,N9Zn2+取代aG1O6,N7Zn2+成为最稳定的配合物.与aG3xMg2+,aG4xMg2+,aG5xMg2+稳定性顺序不同,aG3N1,O6Zn2+和aG4O6,N7Zn2+变成第三和第五稳定的结构,而aG5N1,O6Zn2+变成第四稳定的结构.很显然,除了aG6O6,N7Mg2+与aG6O6,N7Zn2+的稳定性顺序一致都排第六位以外,前五个配合物的稳定性顺序尽管鸟嘌呤异构体相同,结合位点也一样,却因结合离子的不同而导致稳定性顺序都发生重大变化.SI中表S4显示,构成aGnxZn2+稳定性的四个要素除了Es外,其他三项基本变化不大(对各配合物的总能量贡献较小,不影响稳定排序).比较起来,前五个aGnxZn2+(n=1-5)的Es比aGnxMg2+的相应值分别低171.1,204.2,169.2,150.1,167.8 kJ·mol-1.由此可见这是一组比较大但没有规律的数据,这组数据证明锌离子与某一特定异构体结合会“感受”到不同大小的溶剂效应,导致其稳定性顺序不同于相应的aGnxMg2+排序,而且这种“感受”要比aGnxMg2+强烈得多.产生这种现象的原因可能与Zn2+(Ar[3d104s0]2+)和Mg2+(Ne[3s0]2+)不同的电子排布有关.
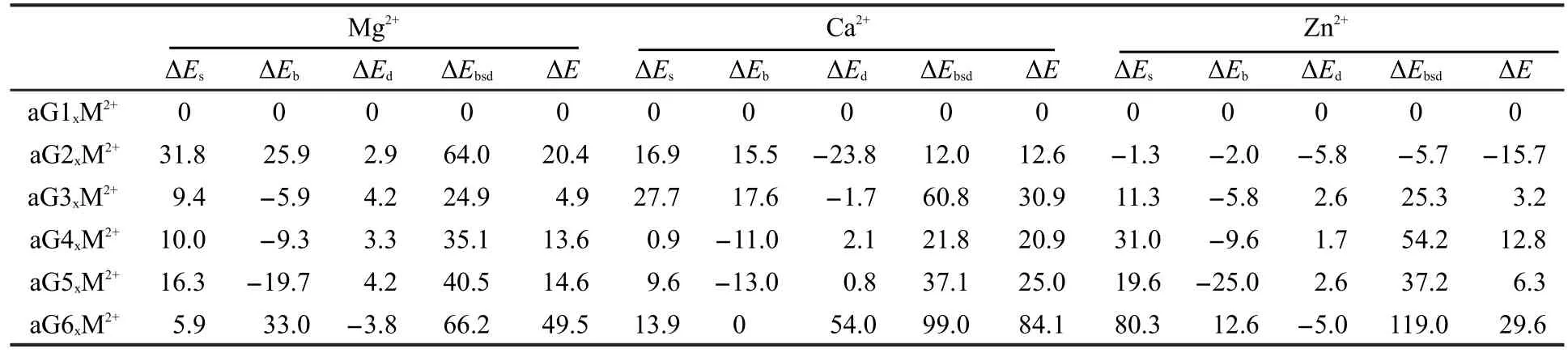
表2 液相中配合物aGnxM2+的各项相对能量Table 2 Various relative energies of complexes aGnxM2+
2.2 气相中的稳定性排序
气相中鸟嘌呤异构体的稳定性顺序为gG2>gG1>gG6>gG8>gG7>gG3>gG9>gG10>gG12>gG4>gG5>gG11>gG13,这一顺序与Hanus[11]和Russo[15]等使用B3LYP/6-311+G**方法得到的结果一致.与液相中的稳定性顺序比较发现,只有前两位的gG1,gG2和最后两位的gG11,gG13的稳定性顺序相同,中间异构体的稳定性都发生了变化.很显然,溶剂效应扮演了很重要的角色.
在气相中gGnxNa+配合物的稳定性的顺序为gG1O6,N7Na+>gG3N1,O6Na+>gG2N3,N9Na+>gG7N3,N9Na+>gG5N1,O6Na+>gG6O6,N7Na+>gG4O6,N7Na+>gG8N1,O6Na+>gG9O6,N7Na+>gG12N2,N3Na+>gG11N7Na+>gG10N3,N9Na+>gG13N3,N9Na+.gGnxK+与gGnxNa+相比,只有gG2N3,N9K+和gG3N1,O6K+,配合物gG5N1,O6K+和gG6O6,N7K+的顺序发生颠倒.这与Kabelac等[12]以MP2方法以及Russo等[15]以B3LYP/6-311+G**所得的稳定顺序一致,不同的是我们得到了gGnxNa+所有可能的配合物并比较了它们的能量,得出了各配合物的稳定性顺序,而且在研究gGnxK+配合物时,补充发现了若干非常稳定的配合物,如排名第二和第六位的gG3N1,O6K+和gG5N1,O6K+.
gGnxCa2+配合物的稳定性顺序为gG1O6,N7Ca2+>gG5N1,O6Ca2+>gG4O6,N7Ca2+>gG3N1,O6Ca2+>gG2N3,N9Ca2+>gG7N3,N9Ca2+>gG6O6,N7Ca2+>gG9O6,N7Ca2+>gG12N2,N3Ca2+= gG12N3,N9Ca2+>gG11N7Ca2+>gG8N1,O6Ca2+>gG13N3,N9Ca2+= gG13N2,N3Ca2+>gG10N3,N9Ca2+.gGnxMg2+配合物中除了gG2N3,N9Mg2+和gG7N3,N9Mg2+的稳定性顺序颠倒外,其余的与gGnxCa2+配合物的基本相同,并且金属离子的结合位点也完全一样.该稳定顺序与Russo[16]在Ca2+/Mg2+与鸟嘌呤异构体(G1,G2,G6,G7,G8)相互作用所得配合物的稳定顺序也完全一致.不同的是我们又补充发现了gG3N1,O6Ca2+/Mg2+,gG4O6,N7Ca2+/ Mg2+,gG5N1,O6Ca2+/Mg2+六种比较稳定的配合物.与gGnxMg2+配合物的稳定性顺序相比,gGnxZn2+中只有三个配合物的顺序发生变化,即gG9O6,N7Zn2+>gG12N3,N9Zn2+>gG6O6,N7Zn2+,这与Kabelac等[12]的结果完全一致,但找到了第六稳定的配合物gG7N3,N9Zn2+.这些稳定异构体的补充对人们了解生物体内可能的离子结合及引起的功能变化至关重要.
比较表S2和表S4发现,gGnxM2+配合物中金属离子与配体的结合强度远大于gGnxM+配合物中结合强度,导致鸟嘌呤异构体发生比较大的形变,并对配合物的稳定性顺序产生重要影响.对于这些结合强度(Eb),Kabelac等[12]利用MP2方法计算的一价金属离子结果与我们的相比,大小及顺序都非常一致.但二价金属离子配合物的结果差别则较大,有的甚至多达100 kJ·mol-1,原因是其采用了MP2方法并对计算结果未采取零点校正所致.而与Russo等[15-16]利用B3LYP/6-311+G**方法计算的数据比较发现,不管对一价还是二价配合物的结合能差值都在20 kJ·mol-1以内,具有合理的一致性,并且我们使用的基组更大,因而结果应该更可靠.
3 结论
采用B3LYP/6-311++G**//6-31+G*方法计算模拟了液(PCM模型)气两相中十三种鸟嘌呤异构体与五种生物体中常见金属离子形成的290个配合物的稳定结构.
系统报道了液相中十三种鸟嘌呤的异构体与五种生物体中常见的金属离子配合物的稳定结构及其稳定性顺序,确定了这些配合物的基态结构: aG1N2,N3Na+/K+,aG1O6,N7Ca2+,aG1N2,N3Mg2+(aG1O6,N7Mg2+), aG2N3,N9Zn2+.并从溶剂-溶质相互作用能、形变能、结合能及异构体的自由能四方面分析了这些金属离子-配合物稳定性差异的原因.结果发现,液相中两个不同一价金属离子与鸟嘌呤异构体形成的配合物的稳定性顺序基本一致,而三个不同二价金属离子配合物的稳定性顺序则表现出不同的规律.而导致它们稳定性顺序不同的原因又各不相同.
同时系统完善了气相中所有金属离子配合物的稳定性排序,并补充报道了很多前人未报道的非常稳定的配合物结构,如gG3N1,O6K+(第三稳定的结构),gG5N1,O6K+(第六稳定配合物),gG3N1,O6Ca2+/Mg2+(第四稳定配合物),gG4O6,N7Ca2+/Mg2+(第三稳定配合物).
简言之,尽管液气两相中GnxM+/2+配合物的稳定性顺序发生了很大的变化,且结合位点也不尽相同,但其最稳定的配合物对应的鸟嘌呤异构体构型都是G1(液相中aG2N3,N9Zn2+除外),这些配合物的基态分别是:aG1N2,N3Na+/K+和gG1O6,N7Na+/K+,a/gG1O6,N7Ca2+, aG1N2,N3Mg2+(aG1O6,N7Mg2+)和gG1O6,N7Mg2+,aG2N3,N9Zn2+和gG1O6,N7Zn2+.
Supporting Information Available: Table S1 lists the relative energies of the complexes aGnxNa+obtained by different models.Various energy values of complexes a/gGnM+and a/gGnxM2+,including solute-solvent interaction energy,binding energy,deformation energy,and relative free energy of the guanine isomers,are given in Tables S2 and S4,respectively. Various values of relative energy of complexes aGnxM+/aGnxM2+based on aG1xM+/aG1xM2+are shown in Tables S3 and S5,respectively.This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
1 Potaman,V.N.;Soyfer,V.N.J.Biomol.Struct.Dyn.,1994,11: 1035
2 Martin,R.B.Acc.Chem.Res.,1985,18:32
3 Muller,J.;Sigel,R.K.O.;Lippert,B.J.Inorg.Biochem.,2000, 79:261
4 Saenger,W.;Cantor,C.R.Principles of nucleic acid structure. New York:Springer,1984
5 Miguel,P.S.;Lax,P.;Willermann,M.Inorg.Chim.Acta,2004, 357:4552
6 Nakano,S.I.;Fujimoto,M.;Hara,H.;Sugimoto,N.Nucleic Acids Res.,1999,27:2957
7 Pettitt,B.M.;Rossky,P.J.J.Phys.Chem.,1986,84:5836
8 Oliva,R.;Cavallo,L.J.Phys.Chem.B,2009,113:1567
9 Sabio,M.;Topiol,S.;Lumma,W.C.J.Phys.Chem.,1990,94: 1366
10 Choi,M.Y.;Miller,R.E.J.Am.Chem.Soc.,2006,128:7320
11 Hanus,M.;Ryjacek,F.;Kabelac,M.J.Am.Chem.Soc.,2003, 125:7678
12 Kabelac,M.;Hobza,P.J.Phys.Chem.B,2006,110:14515
13 Mazzuca,D.;Russo,N.;Toscano,M.J.Phys.Chem.B,2006, 110:8815
14 Pedersen,D.B.;Simard,B.J.Phys.Chem.A,2003,107:6464
15 Russo,N.;Toscano,M.;Grand,A.J.Am.Chem.Soc.,2001,123: 10272
16 Russo,N.J.Phys.Chem.A,2003,107:11533
17 Gresh,N.J.Phys.Chem.B,1999,103:11415
18 Burda,J.V.;Sÿponer,J.;Hobza,P.J.Phys.Chem.,1996,100: 7250
19 Hunter,K.C.;Millen,A.L.;Wetmore,S.D.J.Phys.Chem.B, 2007,111:1858
20 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision C.02.Wallingford,CT:Gaussian Inc.,2004
21 Kumar,A.;Sevilla,M.D.;Suhai,S.J.Phys.Chem.B,2008,112: 5189
22 Ai,H.Q.;Yang,A.B.;Li,Y.G.Acta Phys.-Chim.Sin.,2008,24: 1047 [艾洪奇,杨爱彬,李允刚.物理化学学报,2008,24: 1047]
23 Gustavsson,T.;Bányász,Á.;Lazzarotto,E.J.Am.Chem.Soc., 2005,128:607
24 Shukla,M.K.;Jerzy,L.J.Phys.Chem.A,2005,109:7775
25 Gustavsson,T.;Sarkar,N.;Lazzarotto,E.J.Phys.Chem.B,2006, 110:12843
26 Boys,S.F.Mol.Phys.,1970,19:553
August 13,2010;Revised:September 24,2010;Published on Web:November 3,2010.
Stability of Complexes Combined by Metal Ions(Na+,K+,Ca2+,Mg2+,Zn2+) and Guanine Isomers
ZHAO Yong-Ping AI Hong-Qi∗CHEN Jin-Peng YANG Ai-Bin QI Zhong-Nan
(School of Chemistry and Chemical Engineering,University of Jinan,Jinan 250022,P.R.China)
The order of stability for complexes of differently coordinated metal ions(M+/2+=Na+,K+,Ca2+, Mg2+,Zn2+)with thirteen guanine isomers in gas(g)and aqueous(a)phases was systematically investigated at the B3LYP/6-311++G**level in combination with the polarized continuum model(PCM).Special effort was devoted to differences in the order of stability for aGnxM+/2+(n is the label of guanine isomers,x denotes binding site of M+/2+and guanine isomers)complexes that were obtained in aqueous solutions.An analysis was also performed to determine the reason for these differences with respect to the solute-solvent effect,binding energy,deformation energy,and relative free energy of the guanine isomers. The most stable complexes generated by the five metal ions were:aG1N2,N3Na+,aG1N2,N3K+,aG1O6,N7Ca2+, aG1N2,N3Mg2+(aG1O6,N7Mg2+),and aG2N3,N9Zn2+.The isomer of guanine in the most stable Zn2+complex in the aqueous solution was G2 whereas in the other four most stable complexes it was G1,i.e.,the different active sites in G1 generate the four most stable complexes.Additionally,we report on stable complexes in the gas phase such as gG3N1,O6K+,gG5N1,O6K+,gG3N1,O6Ca2+/Mg2+,and gG4O6,N7Ca2+/Mg2+.
Guanine isomer;Stability;Metal ion;Solute-solvent effect;Binding energy
O641
∗Corresponding author.Email:chm_aihq@ujn.edu.cn;Tel:+86-531-82765475
The project was supported by National Natural Science Foundation of China(20973084,20573047),Natural Science Foundation of Shandong Province,China(Y2008B56)and Research Fund for Young and Middle-aged People of Shandong Province,China(2007BS02009).
国家自然科学基金(20973084,20573047),及山东省自然科学基金(Y2008B56)和优秀中青年科研奖励基金(2007BS02009)资助项目
猜你喜欢
系统仿真技术(2022年4期)2023-01-17 13:01:44
云南化工(2021年8期)2021-12-21 06:37:38
原子与分子物理学报(2021年1期)2021-03-29 07:28:54
国际呼吸杂志(2019年20期)2019-11-23 08:46:08
化工生产与技术(2016年5期)2016-11-07 02:27:43
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
信息记录材料(2016年4期)2016-03-11 15:22:30
合成技术及应用(2015年2期)2016-01-10 10:30:13
