
2苯基-4,5-二(4′-氨基苯基)咪唑的合成*
2010-11-27 11:18张晓云高艳敏
合成化学 2010年3期
吴 伟, 张晓云, 高艳敏
[中国石油大学(华东) 化学化工学院,山东 青岛 266555]
二氨基取代的芳香族化合物是合成聚酰亚胺的重要单体,广泛用于材料合成、粘合剂、耐高温基体树脂等工业领域[1,2]。2-苯基-4,5-二(4′-氨基苯基)咪唑(6)可用于可溶性聚酰亚胺的制备[3]。咪唑类化合物合成一般采用苯偶酰环化的方法, 由于从苯偶酰直接硝化再还原的方法只能得到 3,3′-二取代物[4]。4,4′-二硝基苯偶酰(4)的合成一般通过金属盐催化的苯甲酰氰的偶合反应,其原料的制备和反应过程中使用或产生剧毒的氰化物,另外一种合成苯偶酰的方法是经过片哪醇衍生物的氧化,需使用昂贵的金属铼或铑催化剂进行。作者曾利用对氨基苯甲醛经4合成二氨基取代的洛粉(2,4,5-三苯基咪唑)衍生物[5],但仍存在对氨基苯甲醛不稳定易自身缩合,原料来源受限,且过程中也须使用氰化钾(钠)为催化剂等不足之处。
本文以安息香为原料,经过乙酸酐酯化、乙酸铵噁唑环化、混酸硝化、液溴氧化开环合成了4; 4在乙酸铵/冰乙酸体系中环化生成2-苯基-4,5-二(4′-硝基苯基)咪唑酰(5); 5在三氯化铁存在下经水合肼还原制得6(Scheme 1)。此方法原料来源方便、合成操作简单。
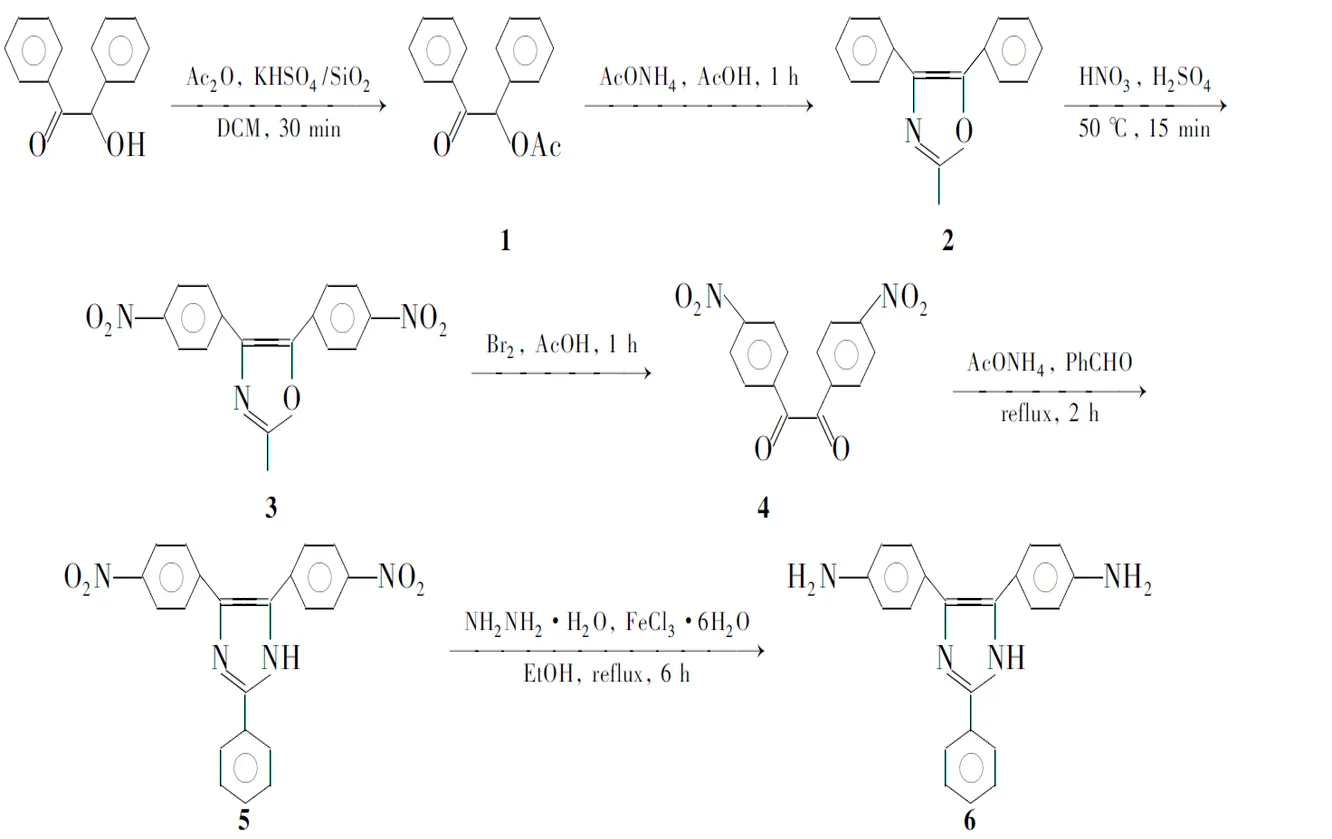
Scheme 1
1 实验部分
1.1 仪器与试剂
厦门大学教学仪器厂显微熔点仪(温度未校正);Bruker DMX 300型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Shimadzu-400型红外光谱仪(KBr压片);Perkin Elmer 2400型元素分析仪。
层析硅胶,试剂级,200目~300目,青岛海洋化工厂分厂;安息香,江苏常州市武进雪堰万寿化工有限公司;其余所用试剂均为国药集团上海化学试剂公司产品或天津化学试剂有限公司产品,使用前未经纯化处理。
1.2 合成
(1) 安息香乙酸酯(1)的合成
将KHSO42 g溶于20 mL水中,加入层析硅胶20 g,搅拌均匀,在烘箱中于120 ℃烘24 h得固体酸酯化催化剂KHSO4/SiO2。
在烧瓶中将安息香42.4 g(200 mmol)溶于二氯甲烷(300 mL)中,加入乙酸酐19 mL(200 mmol) 和KHSO4/SiO220 g,搅拌下于室温反应30 min。过滤,滤饼用甲醇重结晶得无色晶体149.4 g,收率98 %, m.p.81 ℃~83 ℃(81 ℃~82 ℃[6]);1H NMRδ: 7.6~7.2(m, 10H), 6.80(s, 1H), 2.20(s, 3H)。
(2) 2-甲基-4,5-二苯基噁唑(2)的合成
在反应瓶中加入138 g(150 mmol),乙酸铵15 g(200 mmol),冰乙酸40 mL,搅拌下回流反应约1 h(TLC监测)。冷却后倒入碎冰中,用乙醚(4×50 mL)萃取,合并萃取液,用无水硫酸镁干燥过夜,旋除溶剂得无色油状液体235 g,收率99%, b.p.(210~215) ℃/2.6 kPa;1H NMR(CDCl3)δ: 7.65~7.25(m, 10H ), 2.59(s, 3H)。2未经纯化直接用于下一步反应。
(3) 2-甲基-4,5-二(4′-硝基苯基)噁唑(3)的合成
在冰水冷却和充分搅拌下将浓硫酸(28 mL)与浓硝酸(36 mL)混合制得混酸,加热至50 ℃;加入234 g(144 mmol),剧烈搅拌下于50 ℃反应15 min。倾入碎冰中析出黄色固体,过滤,滤饼经空气干燥后用冰乙酸重结晶得亮黄色针状晶体340.7 g,收率 87.0%, m.p.242 ℃~244 ℃;1H NMR(CDCl3)δ: 8.30(d,J=8.5 Hz, 2H), 8.28(d,J=9.0 Hz, 2H), 7.83(d,J=8.5 Hz, 2H), 7.76(d,J=9.0 Hz, 2H), 2.62(s, 3H)。
(4)4的合成
在反应瓶中依次加入液溴20 mL,冰乙酸100 mL,339 g(120 mmol),搅拌下回流反应1 h。倾入碎冰中,析出黄色固体,过滤,滤饼用50%乙酸重结晶得黄色针状晶体432.1 g,收率89%, m.p.212 ℃~214 ℃(212 ℃~213 ℃[7]);1H NMR(CDCl3)δ: 8.40(d,J=9.0 Hz, 2H), 8.22(d,J=9.0 Hz, 2H)。
(5)5的合成
在反应瓶中依次加入冰乙酸250 mL,430 g(100 mmol),乙酸铵100 g(1.3 mol),新蒸苯甲醛21.2 g(200 mmol),搅拌下回流反应2 h。蒸除大部分乙酸,残余物倾入约1 kg碎冰中,析出沉淀,过滤,滤饼水洗、干燥得橙色固体537.4 g,收率97%(经TLC检测未见其它杂质),m.p.>300 ℃;1H NMRδ: 8.45~8.20(m, 8H), 7.35~7.50(m, 5H); IRν: 3 371, 1 602, 1 521, 1 345, 1 200, 1 100, 840, 760 cm-1; Anal.calcd for C21H14N4O4: C 65.28, H 3.65, N 14.50; found C 65.74, H 3.81, N 14.17(元素分析样品经乙酸重结晶 )。
(6)6的合成
在三口烧瓶中依次加入无水乙醇150 mL,515.44 g(40 mmol),活性炭2 g,六水合三氯化铁1 g,搅拌下加热至接近回流,在30 min内滴加80%水合肼15 mL,滴毕,回流反应6 h。趁热过滤,滤液旋转蒸发至干,残余物经混合溶剂[V(甲醇) ∶V(甲苯)=1 ∶5]重结晶得淡黄色固体611.3 g,收率86.7%, m.p.>300 ℃;1HNMRδ: 12.89(s, 1H), 7.48~7.23(m, 5H), 7.22(d,J=9.0 Hz, 4H), 6.60(d,J=9.0 Hz, 4H), 5.05(s, 4H);13C NMRδ: 148.0, 144.6, 130.5, 129.3, 128.2, 127.6, 126.4, 127.3, 125.5, 116.4; IRν: 3 368, 3 210, 1 652, 1 599, 1 515, 1 342, 1 278, 1 181, 1 110, 967, 855 cm-1; Anal.calcd for C21H18N4: C 77.28, H 5.56, N 17.17; found C 77.64, H 5.82, N 16.47。
2 结果与讨论
乙酸酐与安息香的酯化反应传统上主要使用H2SO4作催化剂,副反应较多;近来也有报道使用路易斯酸[如BiNO3, ZrCl4, Zn(ClO4)等]作催化剂。但这些催化剂都存在如反应时间长、转化率较低需使用过量的乙酸酐或试剂来源少等缺点。本文利用固相负载的强酸性盐KHSO4作催化剂,该催化剂易于制备并且避免了使用液体酸需要中和、水洗等后处理过程。结果表明以二氯甲烷或氯仿为溶剂,于室温反应30 min安息香已经完全酯化。反应后只需将溶剂和产生的乙酸蒸除即可得到2。
文献[8]方法合成2使用甲酰胺法或硫脲法,前者试剂较少见而后者使用高沸点的二甲基甲酰胺作为溶剂,后处理较复杂。本文利用乙酸铵作为氨的供体,在冰乙酸中回流使1关环。实验发现增加乙酸铵的用量可以提高收率,缩短反应时间,反应几乎可以定量地完成。
2的硝化反应之关键是反应温度和反应时间(表1)。由于在硝化反应主要阶段反应放热剧烈,因此必须注意充分搅拌使反应物尽可能稳定在一定温度范围内。在50 ℃下利用混酸进行硝化,反应的诱导期很短(数秒)。在反应时间小于10 min时,通过TLC检测在反应混合物中明显见到有部分一硝基产物,但很快进一步被硝化成二硝基产物,该反应在15 min内完成。延长反应时间或提反应高温度均可导致原料的深度硝化,结果得到不溶于乙酸的多硝基产物。
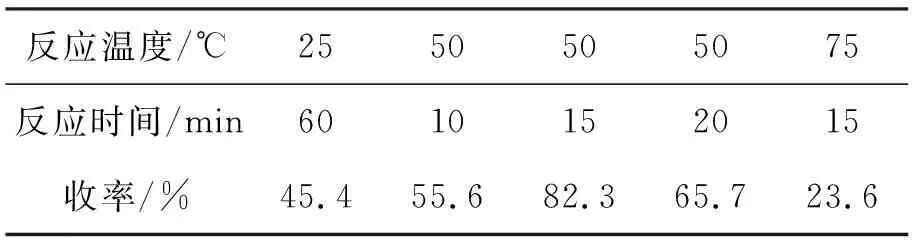
表 1 硝化反应条件对3收率的影响*Table 1 Effect of nitration conditions on the yield of 3
*23.4 g,混酸(H2SO42.8 mL+HNO33.6 mL),其余反应条件同1.2(3)
借助噁唑环将硝基引入苯环的对位后,以溴为氧化剂将噁唑环氧化开环形成苯偶酰结构。此类反应报道较少,本文借鉴了文献[9]的噁唑开环方法。以冰乙酸为溶剂,回流反应约1 h,反应进行顺利,TLC检测未发现有溴代产物。
苯偶酰类化合物在乙酸铵/冰乙酸体系中反应是合成咪唑类衍生物常用的方法。本文未采用乙酸铵和醛大大过量的方法,也能顺利合成5。以水合肼为还原剂,在铁离子催化下,可将5的硝基高效还原为氨基,且操作过程相对简单,单步反应收率达86.7 %。
3结论
(1) 固相负载的酯化催化剂KHSO4/SiO2可高效催化安息香与乙酸酐的酯化反应,反应以二氯甲烷为溶剂在室温下进行完全。得到的安息香乙酸酯在乙酸铵/冰乙酸中回流,以99%收率得到2-甲基-4,5-二苯基噁唑。
(2) 2-甲基-4,5-二苯基噁唑经硝化反应合成2-甲基-4,5-二(4′-硝基苯基)噁唑的适宜条件是:反应温度50 ℃,反应时间15 min。2-甲基-4,5-二(4′-硝基苯基)噁唑可被液溴在冰乙酸中氧化成4,4′-二硝基苯偶酰。
(3) 4,4′-二硝基苯偶酰与苯甲醛在乙酸铵/冰乙酸体系中回流反应生成2-苯基-4,5-二(4′-硝基苯基)咪唑,然后经水合肼还原得到2-苯基-4,5-二(4′-氨基苯基)咪唑,其适宜的还原条件是:催化剂FeCl3·6H2O/活性炭,乙醇为溶剂,回流反应6 h, 2-苯基-4,5-二(4′-氨基苯基)咪唑的总收率63.2%(以安息香计)。
[1] Ishii J, Sunaga T, Nomura M. Organo-soluble polyimides and their applications to photosensitive cover layer materials in flexible printed circuit boards[J].Journal of Photopolymer Science and Technology,2008,21(1):107-112.
[2] Mousa G, Raouf A, Hossein B. Synthesis of soluble and thermally stable polyimide from new diamine bearingN-[4-(9H-carbazol-9-yl)phenyl]formamide pendent group[J].European Polymer Journal,2009,45(11):3108-3115.
[3] 田野,王丽华,丁怀宇,等. 可溶性聚酰亚胺规整多孔膜的形成与控制[J].膜科学与技术,2009,29(4):19-24.
[4] 高妍,张志强,周袭非,等. 间二氨基苯偶酰的合成[J].化学试剂,2005,27(10):627-628.
[5] Wu W, Wang D, Ye C. Synthesis and characterization of addition-type polyimides functionlized with diamine chromophore[J].Polym Bull,1998,41:401-408.
[6] Jeyakumar K, Chand D. Copper perchlorate:Efficient acetylation catalyst under solvent free conditions[J].Journal of Molecular Catalysis A:Chemical,2006,255(1-2):275-282.
[7] Cremlyn R, Swinbourne F, Shode O. Cyclization of benzils[J].Journal of Heterocyclic Chemistry,1987,24(1):117-21.
[8] Asadolah K, Heravi M. Bismuth(Ⅲ) nitrate supported on silica gel,a new catalyst for acetylation of alcohols and phenols under microwave irradiation[J].Phosphorus, Sulfur and Silicon and the Related Elements,2004,179(11):2335-2339.
[9] Hassner A, Fischer B. Synthetic methods.The 4,5- and 2,5-additions to oxazoles[J].Tetrahedron,1989,45(19):6249-6262.
猜你喜欢
食品安全导刊(2020年21期)2020-09-07
农药科学与管理(2019年8期)2019-11-23
农家科技中旬版(2019年9期)2019-10-08
山西农业科学(2019年6期)2019-06-19
山东工业技术(2016年13期)2016-06-29
化学工业与工程(2015年1期)2015-02-10
天津化工(2014年1期)2014-10-22
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年7期)2014-02-28
西南民族大学学报(自然科学版)(2014年6期)2014-02-13
