
聚α-羟基辛酸对W/O乳液电纺聚乳酸纤维毡中阿霉素释放的影响*
2010-11-27 02:30宋宏锐邓英杰陈学思
合成化学 2010年3期
王 浩, 宋宏锐, 邓英杰, 陈学思
(1. 沈阳药科大学 a. 药学院; b. 制药工程学院, 辽宁 沈阳 110016; 2. 中国科学院 长春应用化学研究所 高分子化学与物理国家重点实验室,吉林 长春 130022)
静电纺丝是制备微米甚至纳米级尺度纤维的一种方法,成纤维材料的选择范围很广泛,所制得的纤维可作为无纺布进行应用[1,2]。由于电纺原理是粘性的高分子溶液在高压电场作用下在细针尖部位形成Taylor锥,然后在较远距离的接收板上收集被拉长的细丝,所以该方法形成的纤维直径多在5μm以下,有时甚至在100 nm以下,人们称之为超细纤维。随着医学的进步,超细纤维应用在疾病的预防和治疗上显示良好前景,其可能作为手术后的填充和隔离材料。近十年来,将药物包裹在纤维中作为药物的控制释放载体有越来越多的文献报道[3~5]。
水溶性高分子纤维包裹药物后在水性环境中会很快将药物释放完毕,起不到缓慢控制释放药物的作用所以应用有限[2]。经过嵌段亲水基团或共混改性后,水不溶材料亦可包裹水溶性药物,但仍存在突释行为[4]。为解决此问题,景遐斌等[6]提出乳化纺丝步骤将水溶性药物包裹在水不溶性聚乙二醇-b-聚(L-乳酸)(1, Chart 1)材料管状结构的内腔,有效避免了药物突释,但是蛋白酶K(4)对纤维材料的降解会引起核心部位的药物的耗竭,并且酶的浓度越高,药物耗竭速率越快。在病理状态时,机体的催化酶的生物活性可能会增加很多,比如炎症感染部位的fibrinolysin表达会增多,导致酶活性的增高会使聚酯材料的降解加快,进而加快耗竭药物释放载体中的药物,所以开发一种较耐酶降解的药物释放装置很有必要。

Chart1
最近,Trimaille等[7]合成了聚α-羟基辛酸(PHO), 其结构如同将聚(L-乳酸)(PLLA)的甲基换成正己基,这样会阻碍酶对其酯键的降解。由于己基链的空间阻碍作用,高聚合度PHO较难获得[8], 而分子量较小的PHO由于在溶液浓度较低时粘度很低不能满足成纤维的需要,在电纺时不能拉制成纤维。在PHO中嵌段聚乙二醇(PEG),进行亲水性修饰合成低分子量的聚乙二醇-b-聚(α-羟基辛酸)(2, Chart 1),2作为添加剂进入具有较高分子量1电纺纤维中以改善药物释放控制性能[9,10]。本课题组[11]曾经将较小分子量的2掺入较高分子量的1纤维中,使其在高浓度酶条件下仍能保持纤维形态并能减缓释药速率。
本文采用油包水(W/O)乳化-电纺丝工艺(图1)将阿霉素盐酸盐(3)包裹在纤维中形成核-壳状纤维结构,并且在高浓度蛋白酶K的作用下对其进行降解,考察2是否能改善1对酶降解的耐受性以及是否影响3的释放控制行为。

图 1 W/O乳化-电纺丝工艺示意图Figure 1 Schematic diagram of W/O emulsification-electrospinning technology
1 实验部分
1.1 仪器与试剂
Model XL 30ESEM FEG from Micro FEI Philips型扫描电子显微镜(SEM);Rigaku, D/max 2500V PC型广角X-射线衍射仪(XRD);岛津PU 980型高效液相色谱仪[Dikma Diamonsil C18(4.6 mm×150 mm×5μm),流动相:甲醇-乙腈-0.01 mol·L-1乙酸钠溶液-冰醋酸(40 ∶10 ∶50 ∶1),检测波长:254 nm,流速:1 mL·min-1,柱温:室温,进样量:20μL];UV-975型紫外检测器(UV);Waters泵与Wyatt DAWN EOS(MALLS) DSP(RI)组合凝胶渗透色谱(GPC); BRANSON Digital Sonifier®型空气振荡超声器;静电纺丝装置(浙江东渚涂装仪器厂);HWY-103B型恒温空气浴摇床(智诚分析仪器制造有限公司,上海)。
L-丙交酯,Purac公司;3,6-二己基-1,4-二氧杂-2,5-二酮(辛交酯)按文献[12]方法自制;3,纯度98.7%,浙江海正集团;4(Amesco), Biotechnology Grade;聚乙二醇单甲氧基醚2000(MePEG 2000, Mw=2000)(5), Fluka公司;2-乙基己酸亚锡[Sn(OCt)2],纯度>95%, Sigma公司;十二烷基硫酸钠(SDS)和甲苯,化学纯,天津博迪化工有限公司;其余所用试剂均为分析纯。
1.2 1和2的合成
聚合反应在无水条件下进行,并用氩气充填反应装置。
在圆底烧瓶中加入50.6 g和甲苯50 mL,共沸除水12 h;蒸除剩余甲苯,残余物在乙酸乙酯中重结晶,抽干。加入L-丙交酯30 g,于130 ℃磁力搅拌使其熔融,加入Sn(Oct)27 mg的无水甲苯(1 mL)溶液,开环聚合反应24 h。移入氯仿(1 L)中,搅拌使其完全溶解后倒入冷乙醇中析出白色连续的絮状结晶,挤出有机溶剂和细小残渣;再溶解-析出,重复3次。于40 ℃真空干燥至恒重得分子量分布较窄的1。
在燥圆底烧瓶中加入辛交酯(用乙醚重结晶并真空抽干) 5.0 g,于110 ℃磁力搅拌20 min(挥干残余的水分和有机溶剂),加入5的THF溶液(0.6 g·mL-1) 5.0 mL和Sn(Oct)2的THF溶液(0.205 g·mL-1) 1.5 mL,于100 ℃磁力搅拌快速蒸发THF至熔融状态,开环聚合反应4 h。移入THF(30 mL)中,加入冷冻的正己烷200 mL,搅拌,析出聚合物;离心(3000 转·min-1)分离,白色沉淀用冷正己烷混悬[除去残留的辛交酯和Sn(Oct)2];再离心分离-混悬,重复3次。于40 ℃真空抽干至恒重得白色粉末2。
GPC以氯仿为流动相测定1和2的质均分子量Mw和多分散系数(PDI),1: Mw=8.04×104g·mol-1, PDI=1.22;2: Mw=5.28×103g·mol-1, PDI=1.18。
1.3 乳液静电纺丝
将3200 mg溶解于纯净水(5 mL)中制得水溶液(标记为W-3)作为内水相。
在容量瓶中加入1 1.50 g和CH2Cl210 mL,磁力搅拌12 h(溶液无色、透明、均一);加入SDS 75 mg,搅拌12 h(形成混悬液),用CH2Cl2定容使c(1)=60 mg·mL-1制得有机相(标记为O-1)。加入W-31 mL,磁力搅拌2 h,超声分散制得W/O型乳液,迅速进行静电纺丝制得核-壳状结构[13]纤维毡(标记为F-1),
用类似方法制备纤维毡F-0(1 1.50 g), F-2(1 1.50 g, W-32 mL)和F-3(1 1.50 g,2150 mg, W-32 mL)。
静电纺丝[14]:针头与静电发生器相连,接收屏接地并在其表面包蒙上铝箔,使针头喷丝口与铝箔平面之间的距离即纺丝距离为50 cm,同时使得从直角型针头滴出的乳液速度为2 mL·h-1~3 mL·h-1。开动高压静电发生器至5 000 V,在针头和接收屏之间形成一个高压电场,电纺丝开始。从喷丝口流出的纺丝液在电场力的作用下以高速不规则的螺旋轨迹运行,并被拉伸成为一定形状沉积到接收屏上,在接受屏的后方用电暖气对铝箔上新纺出的成型材料中残余溶剂进行挥发,并确保接收板上的温度为30 ℃左右。揭下接收平板上的橘红色纤维毡,剪成小片备用。
1.4 纤维载药量测定与药物释放曲线绘制
在混合溶剂A[V(DMF) ∶V(CH3Cl)=5 ∶4]中加入3,配制成梯度浓度的标准溶液,UV检测,作药物浓度[c(3)/g·L-1]-吸光度标准曲线(图2)。
将纤维毡(15 mg)溶解在混合溶剂A(10 mL)中,UV测其吸光度,查对图2即可算出纤维毡的载药量(%)(表1)。

c(3)/g·L-1图 2 c(3)-吸光度(UV)标准曲线Figure 2 Standard curve of c(3) and absorbance by UV
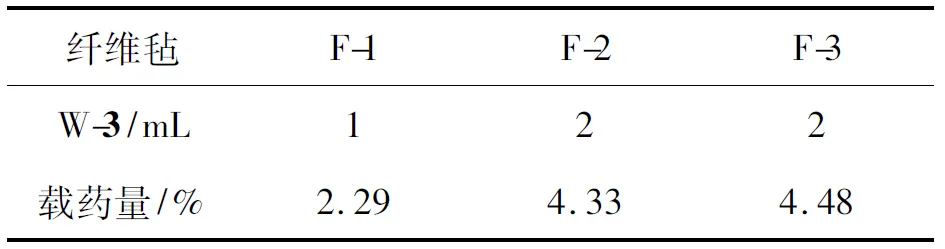
表 1 纤维毡中3的载药量Table 1 The drug loading amount of 3 in fibrofelts
磷酸盐缓冲溶液(PBS, pH 6.8): 将磷酸盐NaH2PO44.35 g和NaH2PO46.75 g溶解在去离子水,定溶至1 L。
将3溶解在PBS中,配制已知梯度浓度的标准溶液。HPLC测定,作c(3)-峰面积标准曲线(图3)。

c(3)/g·L-1图 3 c(3)-峰面积(HPLC)标准曲线Figure 3 Standard curve of c(3) and peak area by HPLC
释放介质: 在PBS中加入4,使4的浓度c(4)分别为2.5μg·mL-1, 5.0μg·mL-1, 10.0μg·mL-1。
将纤维毡[3 cm×2 cm×0.3 mm(15±2 mg)]浸入释放介质(50 mL)中,在预定时间(10 min, 1 h, 6 h, 12 h, 24 h, 48 h, 96 h, 168 h)取出纤维毡,HPLC测定浸过纤维毡的释放介质;取出的纤维毡再浸入新的释放介质(50 mL)中,连续计时,重复操作。通过HPLC所测峰面积查对图3可计算出各时间点的释药量并得出对应的累积释放百分率(Q=释药量/载药量×100%)。
2 结果与讨论
2.1 关于静电纺丝
有机相中的SDS不仅是乳化剂同时也是一种导电物质,所以本实验在静电纺丝过程中不再加入三乙基苄基氯化胺导电。
由于该W/O型乳为热力学不稳定体系,在放置时间过长会出现含药水层逐渐析出在乳液上层的现象,这样单位时间内乳液中乳滴的数目会逐渐减少,进而导致在不同时间段纤维包裹的药物量不均匀,严重时甚至会出现流出针头仅为高分子溶液的现象,所以设定的纺丝速度即注射针头中纺丝液的流出速度要快于单相溶液电纺的速度,使乳液在6 h内纺丝完毕。与文献[6]方法采用氯仿为有机相不同,本文采用CH2Cl2为有机相,由于氯仿比重大于CH2Cl2,乳液静置时分层速度更快,而纺丝时间较长,所以采用分层较慢的CH2Cl2为有机相,并尽可能缩短纺丝时间,以获得载药均匀的纤维毡。
2.2 纤维形态和药物包裹
纤维毡的SEM照片见图4。从图4可以看出,通过W/O乳化-静电纺丝得到的纤维呈交织结构,纤维直径并不均匀,可能是由于内水相小液滴在纺丝的过程中发生了聚合,或是由于W/O型乳剂在纺丝时表面张力变得不均一,从而使得纤维粗细不均。SEM未观察到纤维表面析出颗粒状物,即无3晶体析出。

图 4 纤维毡的SEM照片Figure 4 SEM pictures of fibrofelts

2θ/(°)图 5 样品的XRD谱图Figure 5 XRD spectra of samples
纤维毡的XRD谱图(图5)中未出现3的特征峰,说明乳化-纺丝过后纤维对药物包裹良好。F-1的XRD谱图与F-0相似;F-2在10°~25°有一较大的“馒头峰”说明载药量的增加使得这个角度范围的衍射强度增加;F-3在此衍射角度无F-2的衍射强度,但是在20°~25°有两个小尖峰,说明添加2抵消了“馒头峰” 的出现,而小尖峰可能是由于2掺加进入纤维中后对纤维的表面结晶形态产生了影响。
2.2 纤维中药物的酶降解释放
由表1可知,F-1(内水相体积1 mL)的载药量2.29%, F-2(W-3 2 mL)的载药量4.33%,如果以W-3体积推算,F-2的载药量应为4.58%; F-2的实测载药量低于推算值,原因可能是W/O乳液分层所致,本实验制备的纺丝乳液为热力学不稳定体系,由于纺丝时采用的外相(有机相)为固定体积,当W-3体积由1 mL增加至2 mL时,其体积增多使液滴之间碰撞合并的可能性增加,引起分层;F-3的载药量为4.48,高于F-1和F-2,可能是2的加入可防止W-3液滴合并。
纤维毡的药物释放曲线如图6所示。4对3的释放影响一般在较长的时间才会显现,释放5 min对应的Q可以看成是纤维表面存在的结合不牢固的3的溶解情况,即如果药物在纤维表面有析出,会产生突释现象。实验结果表明,F-1和F-2均未出现明显突释,即无论c(4)是多少,在10 min时都只有很小的色谱峰或几乎检测不到色谱峰,说明1较好包裹了3; F-3在10 min时有突释(Q=10%)。这很有可能是由于2的掺入,使得纤维外壳的PEG链段密度相对较大,在纺丝时PEG可能溶解并在纤维固化后结合一些3,在F-3浸入释放介质时亲水的PEG链段会使药物突释。
在5 min测得的释药量(突释部分)可以作为考察载体对药物包裹状况的指标,F-1和F-2在c(4)=0时,5 min几乎测不到释药量,说明F-1对3有较好的包裹,在c(4)=2.5μg·mL-1时,5 min仅有微量释放。无论c(4)是多少,F-3的释药量均为5%左右,其原因可能为2的亲水链段比例较大,对3产生增溶作用,使其在释放初期溶出量较大。
从总的释放曲线形状来看,2的添加并没有明显地影响F-3中3的释放行为。三种纤维毡在24 h之内的释药速度都较快,几乎达到总载药量的70%。分别来看,24 h后,有4存在的F-1释药量接近100%;c(4)=0时,4天后释药量亦达到80%。对于F-2,c(4)的大小不影响药物释放曲线的趋势,只是在每个时间点,有4存在时,释药量略高;24 h后释药量达80%;4天后达85%。但是我们发现有4存在时,F-2在48 h后几乎降解成细小碎片,由于3本身有一定的亲脂性,其中有一部分已经和纤维碎片中结合在一起沉淀在容器底部,造成没有完全释放的假象。F-3与F-2类似,即c(4)的大小不影响药物释放曲线的趋势,在12 h前,c(4)=10μg·mL-1时,3的释放速度较c(4)=2.5μg·mL-1或0都快,c(4)=2.5μg·mL-1或0的释放量几乎相同,12 h后c(4)=2.5μg·mL-1的释药量反而较c(4)=10μg·mL-1的多,其原因可能是高浓度酶的强烈降解作用使一部分2释放进入PBS中结合了一部分3,或者是由于纤维表面被强烈降解后裸露的2重新吸附了一部分3。

Time/h
超细纤维毡可以看成是固体分散体型药物释放系统,所以其中药物释放可以根据Higuchi方程[15]模拟。采用方程中释放时间的算数平方根t1/2和Q进行线性回归。
尽管本实验中材料对3的包裹类似将药物作为内芯包裹在[1+2]外壳中,但在纤维在固化过程中药物会与高分子进一步地融合而形成固体分散体结构,小分子药物如果分散在高分子载体中,释放用伪Highchi方程来进行拟合后会得到较好结果。
Weibull[16]拟合以lnt为横坐标,以lnln[1/(1-Q)]为纵坐标作图,进行线性回归得方程式和r2值(表2)。Higuchi拟合以t1/2为横坐标,以Q为纵坐标作图,进行线性回归得方程式和r2值(表2)。从表2可以看出,Higuchi拟合的方程斜率说明蛋白酶K的加入对于F-1来说有明显促进药物释放的作用,对于F-2和F-3来说虽然也增加了药物的释放但是斜率差远小于F-1的增加量,说明二倍载药量纺丝得到的纤维中包裹的Dox分子在1中分配比例更高,同时也提示纤维外壁可能由于内水相体积增大而变薄。此外,药物释放曲线也较符合Weibull拟合结果。
F-3在pH 6.8的PBS中降解4天,取出,用去离子水清洗,冻干,观察其表面形态,SEM照片见图7。由图7可见,c(4)=0时,纤维表面仍然光滑;当c(4)=2.5μg·mL-1时,纤维表面出现了小的凸点,这可能是1和2在酶作用下发生了相分离所致;与单相溶液纺丝不同的是,乳化纺丝的纤维在c(4)10μg·mL-1时,纤维表面也有凹凸不平的现象出现,不似单相溶液在高浓度酶的时候表面光滑。
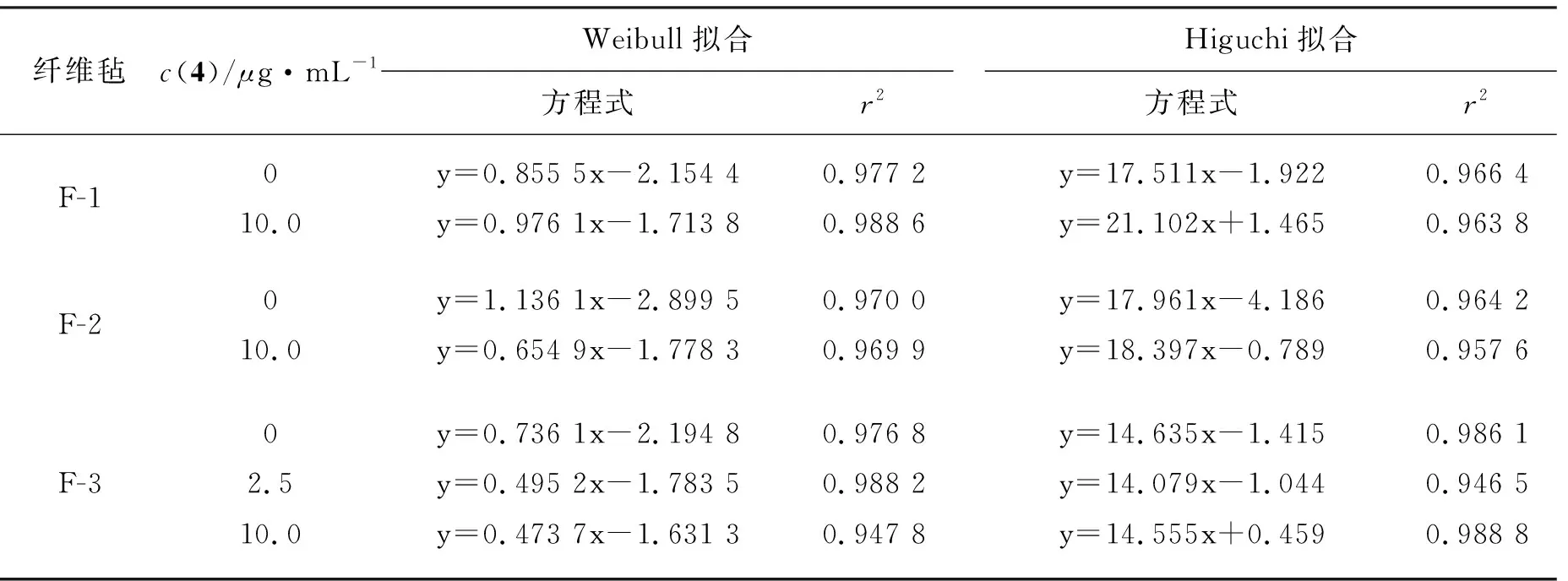
表 2 纤维毡在PBS中的3释放曲线的Higuchi和Weibull拟合*Table 2 Higuchi and Weibull fitting of 3 from fibrofelts in PBS
*根据HPLC测定结果拟合

c(4)=0 μg·mL-1 c(4)=2.5 μg·mL-1 c(4)=10.0 μg·mL-1图 7 在PBS中降解4 d的F-3的SEM照片Figure 7 SEM pictures of F-3 in PBS degradation for 4 days
3 结论
本文采用W/O乳液-静电纺丝法成功制备得到添加聚乙二醇-b-聚(α-羟基辛酸)的内芯含3的聚乙二醇-b-聚(L-乳酸)超细纤维(F-3), F-3具有耐酶降解的特性,在高浓度酶[c(4)=10μg·mL-1]的释放介质中降解7天,纤维仍能保持连接的状态。聚乙二醇-b-聚(α-羟基辛酸)的掺入使得纤维毡耐受高浓度酶的降解,且对3的释放行为几乎没有影响。将分子量较小的非降解性聚合物掺入到可生物降解的分子量较大的高分子中进行纺丝后所得到的纤维便有耐受酶降解的作用,并且对包裹在其中的药物释放行为不产生影响。而不必将该种分子嵌段到形成载体材料的主要高分子中,因为对材料进行改性时,物理掺杂相对于化学聚合反应来说毕竟较为简单。此实验现象提示并再一此证明聚α-羟基辛酸对酶的降解有耐受作用,如果将辛交酯中的一个羟基辛酸置换成乳酸或乙醇酸再开环聚合制备得到的聚合物,其降解性能又将如何,其作为掺杂材料会有什么新的应用价值等是值得进一步研究的。
[1] Kim K, Luu Y K, Chang C,etal. Incorporation and controlled release of a hydrophilic antibiotic using poly(lactide-co-glycolide)-based eletrospun nanofibrous scaffolds[J].Journal of Controlled Release,2004,(98):47-56.
[2] 陈辰,曹传宝,马西兰,等. 静电纺丝丝素蛋白水溶液制备无纺布[J].北京生物医学工程,2007,26(1):10-12.
[3] Xu X, Chen X, Xu X ,etal. BCNU-loaded PEG-PLLA ultrafine fibers and their in vitro antitumor activity against Glioma C6 cells[J].Journal of Controlled Release,2006,114:307-316.
[4] Luu Y K, Kim K, Hsiao B S,etal. Development of a nanostructured DNA delivery scaffold via electrospinning of PLGA and PLA-PEG block copolymers[J].Journal of Controlled Release ,2003,89:341-353.
[5] Jiang H L, Hu Y Q, Li Y,etal. A facile technique to prepare biodegradable coaxial electrospun nanofibers for controlled release of bioactive agents[J].Journal of Controlled Release,2005,108:237-243.
[6] Xu X L, Yang L X, Xu X Y,etal. Ultrafine medicated fibers electrospun from W/O emulsions[J].Journal of Controlled Release,2005,108:33-42.
[7] Thomas Trimaille, Karine Mondon, Robert Gurny,etal. Novel polymeric micelles for hydrophobic drug delivery based on biodegradable poly(hexyl-substituted lactides)[J].International Journal of Pharmaceutics,2006,319(1-2):147-154.
[8] Thomas Trimaille, Michael Möller, Robert Gurny. Synthesis and ring-openning polymerization of new monoalkyl-substituted lactides[J].J Polym Sci: Pol Chem,2004,42:4379-4391.
[9] Morita T, Horikiri Y, Suzuki T,etal. Applicability of various amphiphilic polymers to the modification of protein release kinetics from biodegradable reservoir-type microspheres[J].European Journal of Pharmaceutical and Biopharmaceutics,2001,51:45-53.
[10] Kenawy E R, Bowlin G L, Mansfield K,etal. Release of tetracycline hydrochloride from electrospun poly(ethylene-co-vinylacetate),poly(lactic acid),and a blend[J].Journal of Controlled Release,2002,81:57-64.
[11] Hao Wang, Hongrui Song, Xuesi Chen,etal. Ibuprofen release from PEG-PLLA electrospun fibers containing PEG blocked polyα-hydroxyl octanoic acid as an additive[J].Chinese Journal of Polymer Science,2010,28(3):417-425.
[12] 王浩,邓英杰,王玉玲,等.α-羟基辛酸及其交酯的合成工艺改进[J].合成化学,2008,16(1):53-55.
[13] Xu X L, Chen X S, Jing X B,etal. The release behavior of doxorubicin hydrochloride from medicated fibers prepared by emulsion-electrospinning[J].European Journal of Pharmaceutics and Biopharmaceutics,2008,70:165-170.
[14] 董存海,段斌,袁晓燕,等. 静电纺丝制备聚丙交酯超细纤维[J].生物医学工程学杂志,2005,22(6):1245-1248.
[15] 童珊珊,余江南,徐希明,等. 反相高效液相色谱法测定阿霉素在小鼠血清中的含量[J].江苏大学学报(医学版),2002,12(6):563-565.
[16] 苏德森,王思玲. 物理药剂学[M].北京:化学工业出版社,2004.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
小哥白尼(趣味科学)(2022年2期)2022-05-25
云南化工(2021年7期)2021-12-21
环境卫生工程(2021年4期)2021-10-13
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
孩子(2020年11期)2020-11-17
趣味(数学)(2019年6期)2019-10-17
科学大众·小诺贝尔(2016年11期)2017-01-10
海峡科技与产业(2016年3期)2016-05-17
燕山大学学报(2015年4期)2015-12-25
