
不同地理种群栗瘿蜂的mtDNA的16S rRNA基因部分序列及其系统进化关系分析
2010-11-26 08:38:42贺一原朱道弘
湖南师范大学自然科学学报 2010年3期
范 斌,贺一原,朱道弘,2
(1.中南林业科技大学昆虫行为与进化生态学实验室,中国 长沙 410004;2.湖南第一师范学院,中国 长沙 410205)
栗瘿蜂(DryocosmuskuriphilusYasumatsu)属膜翅目(Hymenoptera)瘿蜂科(Cynipidea)栗瘿蜂属,每年发生1代,主要危害板栗(Castaneamollissima),也危害茅栗(C.sequinii)和锥栗(C.henryi).被害枝不能抽发新梢,严重时枯死,不仅影响当年生长和结果,而且影响次年的产量,是危害我国栗树生长和结果的主要害虫之一.该虫分布于陕西、湖北、湖南、福建等我国大部分板栗产区,日本、朝鲜等国也有报道[1-3].
线粒体DNA(mitochondrial DNA, mtDNA)是核外DNA,由于它具有分子结构稳定,母性遗传、替换速率相对快及缺少重组等特征,已成为昆虫分子系统学常被选择的研究对象[4].对于栗瘿蜂而言,因为其特殊的生殖机制(孤雌生殖),使得栗瘿蜂线粒体DNA成为其进化研究中一种有用的分子标记.而16S rRNA基因又是线粒体上进化速率中等的基因,适合于较高分类单元的系统发育进化的研究.目前,国内对栗瘿蜂研究主要集中在生活史、发生规律和防治策略等方面,而分子水平研究尚未见报道.本研究以栗瘿蜂线粒体内16S rRNA基因作为分子标记,对福建福州、湖南怀化、山东泰安、河北保定、湖南浏阳、广西桂林6个栗瘿蜂地理种群的线粒体DNA进行了序列测定和分析,为我国栗瘿蜂的分子进化提供理论依据.
1 材料和方法
1.1 材料
实验所用栗瘿蜂的虫瘿分别于2005年7月采自福建福州、湖南怀化、山东泰安、河北保定,2009年7月采自湖南浏阳、广西桂林.采集的虫瘿于实验室置于养虫笼(40 cm×40 cm×20 cm)内保存,成虫羽化当日收集的成虫以无水乙醇浸泡,于-20 ℃的冰箱保存备用.
1.2 样品DNA的提取
栗瘿蜂1个体置于装有100 μL STE 缓冲液(100 mmol/L NaCl,10 mmol/L Tris-HCl,1 mmol/L EDTA,pH 8.0)的Eppordef管(1.5 mL)内,将其充分捣碎.加质量分数为10%的SDS溶液10 μL,并加2 μL蛋白质分解酶K(20 g/L),以快速混匀器(XL96-B)混匀,置于电热恒温水浴锅(DK-98-1)内,37 ℃水浴30 min.加100 μL PCL (苯酚、氯仿、异戊醇体积比为 25∶24∶1)进行抽提,台式离心机(TG16-WS)离心10 min,转速为5000 r/min(下同);提取上清液,再加100 μL PCL二次抽提,并二次离心(10 min).离心后的抽提液加入10 μL NaAc(3 mol/L)和250 μL无水乙醇,-20 ℃过夜.过夜后的样品以落地式冷冻离心机(J-25)低温(5 ℃)离心20 min,转速为14 000 r/min(下同);去上清液,用体积分数为75 %的冷无水乙醇洗涤后再低温离心20 min,去上清液并常温干燥.所得的DNA样品加50 μL TE 缓冲液,于4 ℃过夜,-20 ℃保存备用.
1.3 PCR扩增
PCR扩增的上游、下游引物分别为:
5-TRACTGTRCAAAGGTAGC-3;
5-TTAATTCAACATCGAGGTC-3[5](上海生工生物有限公司合成)
PCR反应体系为50 μL,其中10×Buffer(10 mmol/L)5 μL,dNTP(10 mmol/L)1 μL,上下引物(10 μmol/L)各1 μL,Taq 酶(2.5 mol·min-1·L-1)1 μL,DNA 1 μL,双蒸水补足50 μL.扩增条件为96 ℃预变性3 min,接着进行35个热循环:96 ℃变性15 s,49 ℃退火15 s,72 ℃延伸15 s,循环完成后在72 ℃延伸10 min.扩增产物用质量分数为1.2%的琼脂糖凝胶于0.5×TBE 缓冲液中电泳,电压70 V,电泳时间约1 h.电泳后以溴化乙锭(EB)染色30 min,经凝胶成像系统检测并拍照.DNA 分子量采用佳和生物科技有限公司的Mark DL2000标记.每次PCR扩增均设双蒸水的阴性对照.
1.4 16S rRNA基因序列测定和分析
扩增产物经凝胶成像系统检测后,将PCR产物送北京天根生化科技有限公司纯化回收,进行双向测序,以确保序列的可靠性.使用Clustal X 1.81软件对所得栗瘿蜂线粒体16S rRNA基因序列进行排列比对,然后将序列在NCBI中用BLAST进行相似性搜索,确定扩增的片段为目标基因序列.利用Mega 4.0中的双参数模型计算各种群的遗传距离,以落叶松种子小蜂(EurytomalaricisYano)为外群,并用邻近法(NJ),最小进化法(ME),非加权配对算术平均法(UPGMAN)重建系统进化树,系统进化树中结点的自举检验置信度以1 000次自导复制计算估计.
2 结果
2.1 PCR扩增结果
从图1可见,栗瘿蜂16S rRNA的PCR扩增产物经凝胶电泳检测后,大约在430~470 bp之间,为实验扩增的目的基因范围,且条带清晰无杂带,选取亮而且清晰的条带纯化后测序.
2.2 6个不同地理种群的栗瘿蜂16S rRNA基因序列比较
经Mega4.0软件分析得出,保定种群、泰安种群、浏阳种群、福州种群、桂林种群、怀化种群这6个种群的差异非常明显,碱基的缺失、插入、转换和颠换现象严重.
2.3 6个不同地理种群的栗瘿蜂16S rRNA基因序列组成分析
综合从NCBI检索获得的其他栗瘿蜂科中枝跗瘿蜂(Ibalia)的16S rRNA基因序列进行比对,同源性达80%以上,说明所得序列为栗瘿蜂科16S rRNA基因序列.利用Mega 4.0软件统计出这6个不同地理种群的栗瘿蜂16S rRNA基因序列间变异位点和A、T、C、G的平均含量.其中,保守位点(C)88个,变异位点(V)270个,简约信息位点(Pi)155个,自裔位点(Si)15个,分别占24.6%、75.4%、43.3%、32.1%.T、C、A、G碱基平均含量分别为42.6%、8.1%、41.5%、7.8%,其中(A+T)含量(84.1%)明显高于(C+G)含量(15.9%).
2.4 6个不同地理种群的栗瘿蜂16S rRNA基因碱基转换/颠换数及遗传分析
根据16S rRNA基因序列,通过Mega4.0软件分析出6个不同地理种群的栗瘿蜂的遗传距离为0.168 4~0.538 7,平均遗传距离为0.398 0.由表1可以看出桂林和怀化种群间的遗传距离最小(0.168 4),亲缘关系最近;浏阳和桂林种群间的遗传距离最大(0.538 7),亲缘关系最远.整个部分序列中,核苷酸的转换/颠换(TS/TV)平均值为0.4,替换以颠换为主,并且各序列变异位点颠换多于转换(表1).
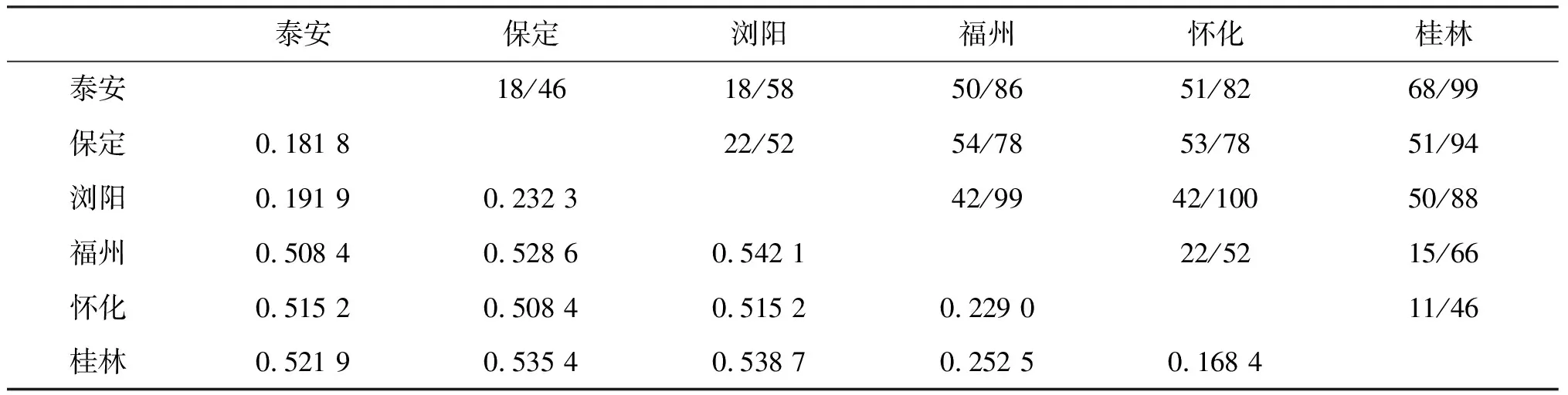
表1 目的基因16S rRNA的序列转换/颠换数与遗传距离
注:对角线上三角的数据为转换/颠换数,对角线下三角的数据为遗传距离.
当转换/颠换比值小于2.0时,此基因序列突变达到饱和状态,受进化噪音的影响可能性较大[6].本研究中的6个地理种群的转换/颠换比平均值为0.4,说明栗瘿蜂的突变已经达到了饱和状态,在重建系统树时受饱和效应的影响也越大.为了避免碱基替换饱和分析对系统进化树分析的影响,本文以遗传距离为横坐标,转换数和颠换数为纵坐标进行散点分析(图2).分析结果显示,颠换的数值明显高于转换的数值,随着遗传距离的增大,颠换数增加的速度大于转换数,且转换数趋于稳定,而颠换数呈直线上升的趋势.转换和颠换的比率随着遗传距离的增加也并未出现饱和状态,而是呈稳定性的线性增长,因此,在分析中将所有的转换和颠换信息应用于系统进化树的构建.
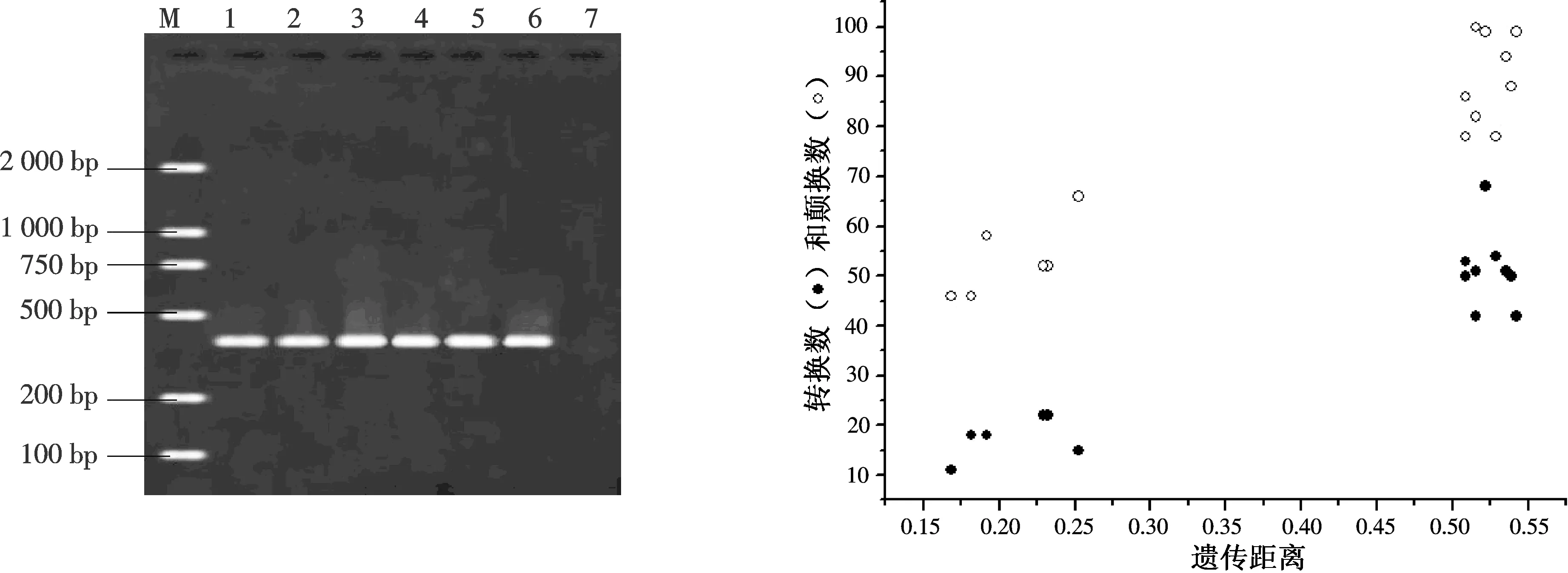
M:Marker2000;1:保定;2:泰安;3:浏阳;4:怀化; 5:福州;6:桂林;7:阴性对照图1 栗瘿蜂6地理种群16S rRNA基因的PCR扩增 图2 栗瘿蜂6地理种群碱基替换饱和分析
2.5 6个不同地理种群的栗瘿蜂16S rRNA分子系统进化树
利用Mega 4.0中的双参数模型分析,以落叶松种子小蜂(EurytomalaricisYano)为外群,用邻近法(NJ法),最小进化法(ME),非加权配对算术平均法(UPGMAN)重建系统进化树,系统进化树中结点的自举检验置信度以1 000次重复估计计算 (图3~图5).
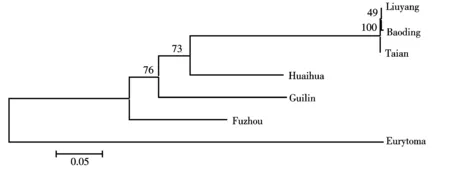
图3 基于16S rRNA基因序列构建不同地理种群的栗瘿蜂的NJ树
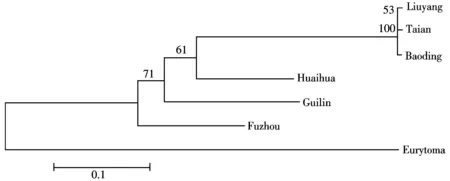
图4 基于16S rRNA基因序列构建不同地理种群的栗瘿蜂的ME树
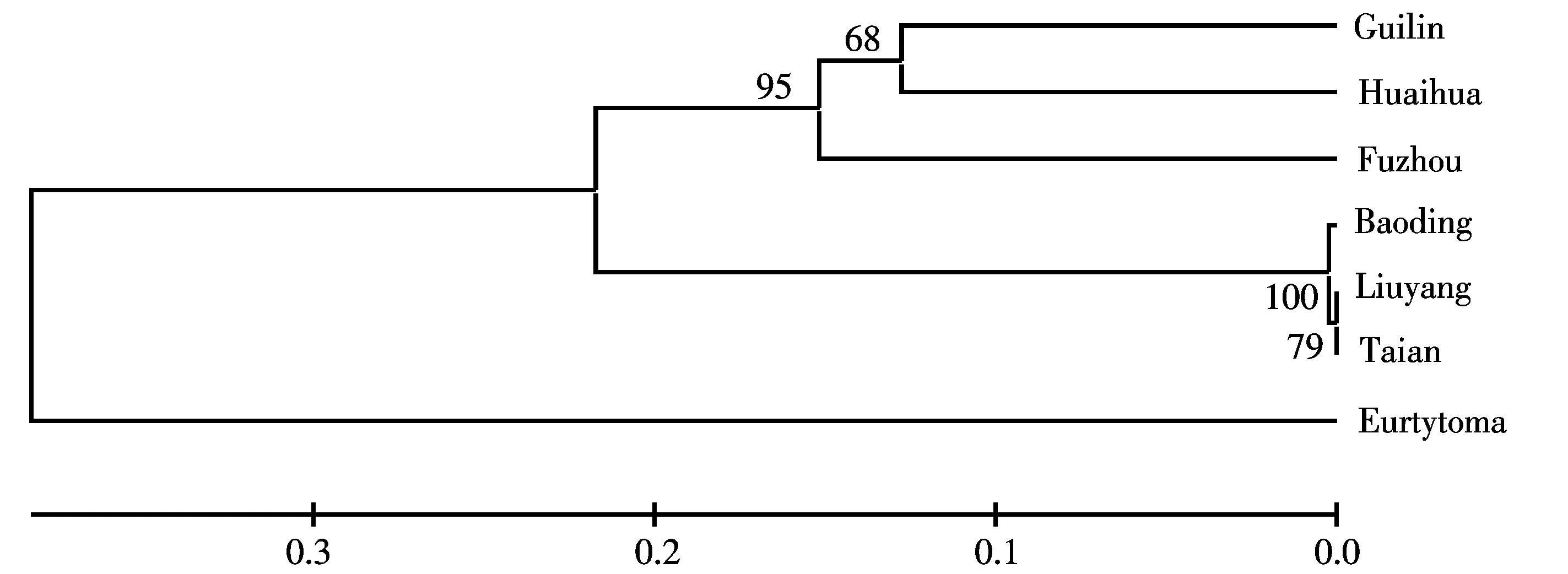
图5 基于16S rRNA基因序列构建不同地理种群的栗瘿蜂的UPGMAN树
利用NJ、ME、UPGMAN分析得到的分子系统发育树拓扑结构基本一致,6个不同地理种群大致可以分为两个支系,第一个支系由保定、浏阳、泰安种群组成,并有着极高的置信度(100%),说明这三者之间具有较近的亲缘关系;第二个支系由福州、桂林、怀化种群组成,前期已经出现分化,且置信度相对较低.从总体上看,这6个地理种群在聚类上呈现出一种平行式的分布关系,并没有因为地理环境的远近而显示出相应的差异性,如空间位置相距较远的浏阳和泰安种群并不一定存在大的差异,而较近的怀化和浏阳种群的差异反而更加显著.
3 结论与讨论
mtDNA 16S rRNA既具有保守性,又具有高变性.保守性能够反映出物种的亲缘关系,为系统发育重建提供线索,高变性则能揭示出物种的特征核苷酸序列,是属种鉴定的分子基础.目前国内外对昆虫16S rRNA的报道较多,但对栗瘿蜂的16S rRNA研究还尚未见报道,而对栗瘿蜂的研究又主要集中在对其生物学特性、发生和防治等方面,如任淑艳[7],黄汉杰[8]等;在分子水平上的研究也仅见于贺一原[1-2]和朱道弘[1-2]对栗瘿蜂的Wolbachia感染研究.国外对瘿蜂科的其他种研究较多,如Dowton M.[9]对枝跗瘿蜂(Ibalia)16S rRNA的研究,Buennige M.[10]对食榕小蜂(Sycophila)的研究等.本研究对我国栗瘿蜂线粒体DNA 16S rRNA基因序列测定和分析,在一定程度上能为栗瘿蜂的系统演化提供分子生物学方面上的证据.
对基因序列进行分析,结果表明,T、C、A、G碱基平均含量分别为42.6%、8.1%、41.5%、7.8%,其中(A+T)含量(84.1%)明显高于(C+G)含量(15.9%),表现出明显的A+T碱基偏嗜,且A和T碱基含量相当,这与Simon[11]等报道的昆虫线粒体基因的碱基组成特征基本一致.本文所示的6个不同地理种群的栗瘿蜂16S rRNA基因片段中具有较多的转换、颠换、插入和缺失现象,这可能是由于16S rRNA基因是一非编码蛋白基因,在进化中所受到的选择压力相对较小的缘故[12].
尽管建树方法的设置参数不同,但是,这6个不同地理种群的分化趋势大致相同,即:泰安、保定、浏阳种群亲缘关系较近,福州、桂林、怀化种群分化程度较高.分化原因可能与栗瘿蜂的生活习性相关,因栗瘿蜂的迁飞能力弱,很难远距离的转移,在地理隔离的作用下,栗瘿蜂由于寄主、气候等外界环境因素的影响,向着适应自己环境的方向进化.另一方面,种群突变和遗传漂变也是导致遗传距离差异的重要因素.
系统拓扑树上的有些分支的置信度不是太高,许多低于70,如NJ上的保定种群和浏阳种群甚至低到49,其原因可能是所研究的序列片段太短(450左右),不能提供足够的遗传信息[13].另外,整个实验过程中所选择的地理种群少,并没有完全反映出我国栗瘿蜂不同地理种群的遗传分化特征,因此对我国不同地理种群的栗瘿蜂遗传分化还有待进一步探讨.
参考文献:
[1] 贺一原,朱道弘,赵吕权. ftsZ基因和16S rDNA特异引物检测栗瘿蜂体内Wolbachia感染研究[J]. 湖南师范大学自然科学学报,2007,30(2):113-115.
[2] 石淑英. 栗瘿蜂发生规律及防治技术研究进展[J]. 北方园艺,1997,5(5):29-30.
[3] 朱道弘,贺一原,赵吕权,等. 栗瘿蜂体内Wolbachia的感染及其wsp基因序列分析[J].林业科学,2007,43(3):133-137.
[4] 黄 原.分子系统学——原理、方法及利用[M]. 北京:中国农业出版社,1998.
[5] SCHULMEISTER S,WHEELER W C,CARPENTER J M. SimμLtaneous analyis of the basal lineages of Hymenoptera (Insecta) using sensitivity analysis[J]. Cladistics,2002,18(3):455-484.
[6] KNIGHT A,MINDELL D P. Substitution bias, weighting of DNA sequence evolution, and the phylogentic position of fea, sviper[J]. Sys Bio,1994,42(1):18-31.
[7] 任淑艳. 栗瘿蜂的发生与防治[J]. 保定果树,2000,3(3):57-58.
[8] 黄汉杰,陈炳旭,孙姒纫,等. 栗瘿蜂的发生与防治[J]. 中国果树,1998,2(2):33-36.
[9] DOWTON M,AUSTIN A D. Molecular phylogeny of the insect order Hymenoptera:apocritan relationships[J]. Nat Acad Sci,1994,91 (21):911-915.
[10] BUENNIGE M,HILKER M,DOBLER S. A molecμLar phylogeny of eurytomid wasps inferred from DNA sequence data of 28S,18S,16S and COI genes[J]. Mol Phylogenet Evol,2004,31(1):300-307.
[11] SIMON C,FRQTI F,BECKENBACH A,etal. Evolution,weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved PCR primers[J]. Ann Entomol Soc Am,1994,87(5):651-701.
[12] 蒋国芳,陈爱辉,冯金叶. 九种蟋蟀mtDNA-16S rRNA 基因序列及其系统进化[J]. 昆虫分类学报,2004,26(2):249-254.
[13] 王玉江,高天翔. 中国和越南青蟹线粒体16S rRNA 基因序列分析[J]. 中国海洋大学学报,2005,35(4):554-558.
猜你喜欢
湘潮(上半月)(2023年6期)2023-08-11 04:05:32
幼儿画刊(2023年6期)2023-07-18 07:02:00
猪业科学(2022年10期)2022-11-03 09:45:54
天津市教科院学报(2021年5期)2021-11-10 07:32:40
生物学通报(2021年9期)2021-07-01 03:24:44
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
饮食保健(2018年16期)2018-08-17 01:58:30
