
HPLC法同时测定复方甘草酸苷片中甘氨酸和蛋氨酸的含量
2010-11-17 08:31吴小曼石蓓佳
中国生化药物杂志 2010年6期
吴小曼,徐 滟,石蓓佳
(1.江苏省食品药品检验所,江苏 南京 210008;2.南京中医药大学,江苏 南京 210046)
复方甘草酸苷片是由甘草酸苷、甘氨酸和蛋氨酸组成的复方制剂,为一安全、有效的保肝护肝制剂,具有保护肝细胞膜、抗炎、免疫调节等作用。本品按进口药品复核标准[1]检验,其中苷氨酸采用薄层扫描法测定,蛋氨酸采用回滴定法测定,操作步骤较为繁琐。有文献报道采用2,4-二硝基氟苯衍生化[2-3]或氯甲酸 9-芴基甲酯衍生化[4-5]HPLC法同时测定两者含量,但衍生化法影响因素较多。采用HPLC法直接测定本品中两种氨基酸含量的方法尚未见报道,本文参照有关文献[6]建立了HPLC法直接测定复方甘草酸苷片中甘氨酸和蛋氨酸的含量。
1 仪器与试药
Agilent 1100高效液相色谱仪(包括二元输液泵、自动进样器、柱温箱、DAD检测器)。
甘氨酸对照品(批号:140689-200401)、蛋氨酸对照品(批号:140684-200401),中国药品生物制品检定所;复方甘草酸苷片,江苏某药业有限公司,批号 :08086A、08086B、08086C;乙腈为色谱醇;水为纯化水;其他试剂为分析纯。
2 方法与结果
2.1 色谱条件
采用Agilent HC-C18(2)色谱柱(4.6 mm×250 mm,5μm);以乙腈 -0.05 moL/L磷酸二氢钾溶液(50%磷酸调节 p H值至2.5)(5∶95)为流动相;流速:0.8mL/min;检测波长:200 nm;柱温:30℃,进样量:10μL。
2.2 溶液的制备
对照品溶液:精密称取甘氨酸与蛋氨酸对照品适量,加水溶解并稀释制成每1mL约含甘氨酸与蛋氨酸各 0.5mg的溶液,摇匀,即得。
供试品溶液:取本品 20片,精密称定,研细,精密称取适量(约相当于甘氨酸和蛋氨酸各 25mg),置50mL量瓶中,加水溶解并稀释至刻度,摇匀,滤过,取续滤液。
2.3 专属性试验
对照品溶液和供试品溶液的色谱图见图1。另按处方比例配制不含甘氨酸与蛋氨酸的阴性样品,再按供试品溶液的制备方法制备得阴性对照溶液,进样,记录色谱图(图1);另取本品细粉适量(约相当于甘氨酸和蛋氨酸各 25mg),置50mL量瓶中,加水溶解并稀释至刻度,摇匀,滤过,取续滤液进样,利用DAD检测器进行色谱峰纯度检查,记录色谱图,甘氨酸峰的纯度因子为997.349,蛋氨酸峰的纯度因子为997.788。结果均表明,处方中的辅料及甘草酸苷对测定无干扰。
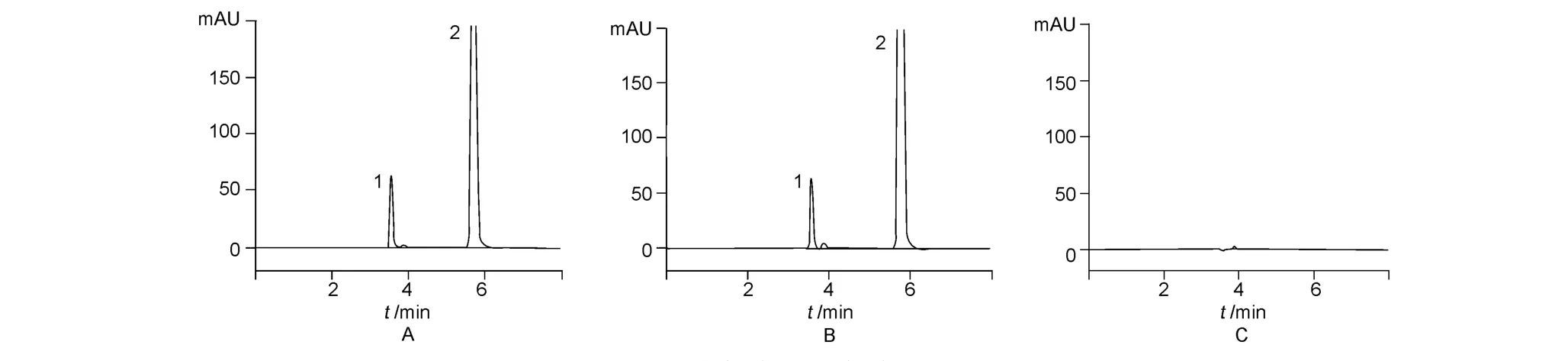
图1 对照品(A)、供试品的(B)及阴性对照(C)的HPLC色谱图Fig.1 HPLC chromatograms of reference substance(A),sample(B)and blank sample(C)
2.4 线性关系
取甘氨酸与蛋氨酸对照品各约40mg,精密称定,置20mL量瓶中,加水溶解并稀释至刻度,摇匀。精密量取0.5,1.0,2.0,4.0,5.0mL,分别置10mL量瓶中,加水稀释至刻度,摇匀,精密量取10μL,分别注入色谱仪,记录色谱图,以峰面积(A)对浓度(C)作线性回归,得回归方程为:甘氨酸 A=667.21 C-6.072 9,r=1.000 0;蛋氨酸 A=9 255.1 C+122.9,r=0.999 9。
结果表明,在0.10~1.0mg/mL范围内,甘氨酸与蛋氨酸溶液浓度均与峰面积呈良好线性关系。
2.5 回收率试验
取已知含量的本品细粉适量(约相当于甘氨酸和蛋氨酸各 12.5mg),分别置9个 50mL量瓶中,再分别精密称取甘氨酸与蛋氨酸对照品各 10,12.5和15mg,各 3份,置上述量瓶中,加水溶解并稀释至刻度,摇匀,取续滤液作为供试品溶液,测定,结果见表1。甘氨酸和蛋氨酸的平均回收率分别为100.29%和99.58%,RSD(n=9)分别为0.97%和0.75%。
2.6 溶液的稳定性
取供试品溶液自配制后分别在0,2,4,6,8 h进样,测定峰面积,甘氨酸与蛋氨酸的RSD分为0.54%与0.34%,表明该溶液在8 h内稳定。
2.7 重复性试验
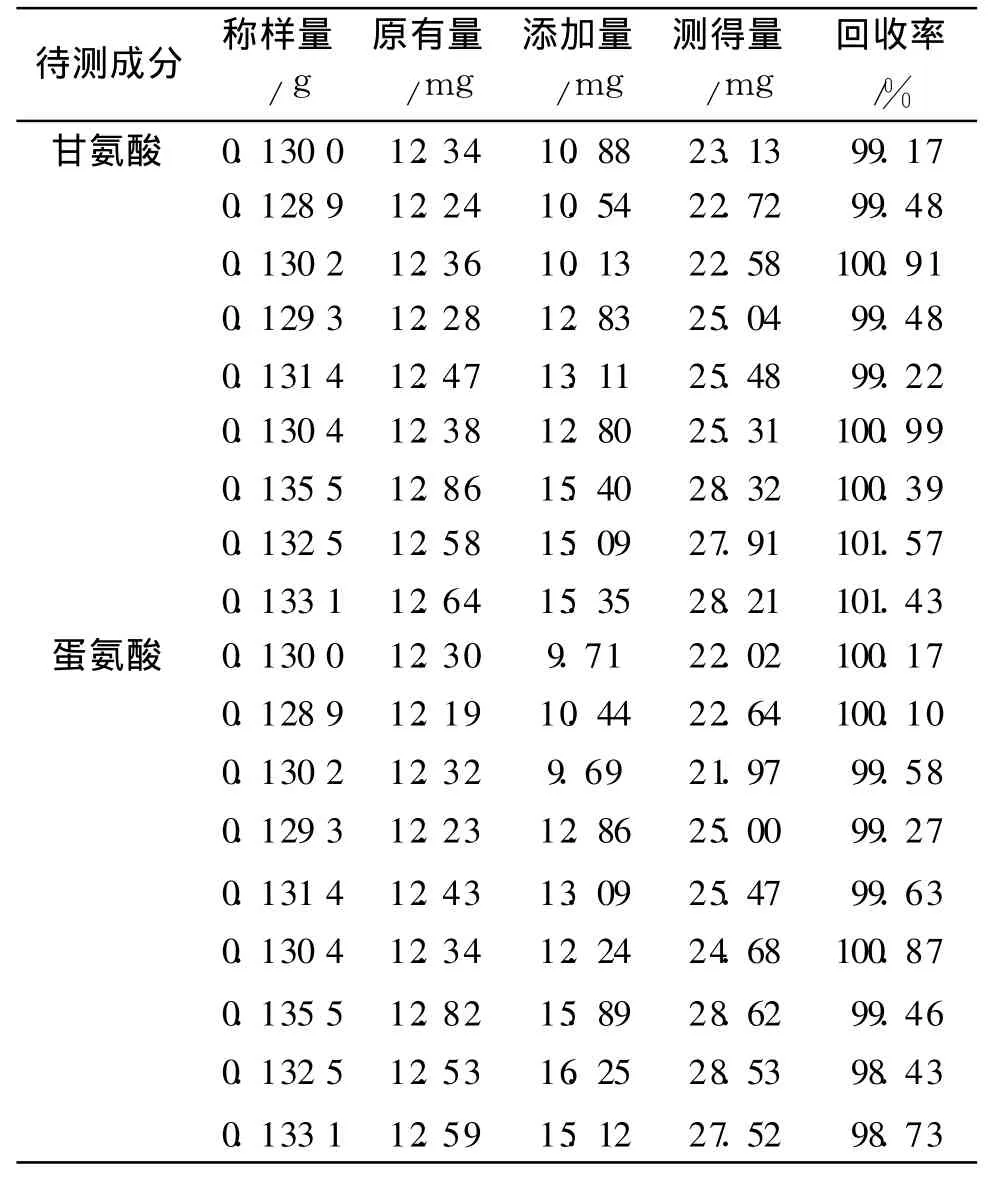
表1 甘氨酸与蛋氨酸的加样回收率试验(n=9)Tab.1 Recovery rate of glycine and methionine(n=9)
取同一批样品(080806A),同法配制 6份供试品溶液,分别测定含量。甘氨酸和蛋氨酸的平均含量分别为100.4%和100.0%,RSD分别为0.53%和0.76%。表明方法的重复性良好。
2.8 含量测定
本品 3批,分别按 2.2项下的方法制备对照品溶液及供试品溶液,按 2.1项下的色谱条件,进样10μL,记录色谱图,按外标法分别以峰面积计算含量,同时与文献方法[1]测定结果比较,见表2,甘氨酸的测定结果有较大差异,这可能与薄层扫描法精密度与准确度都不高有关,两种方法蛋氨酸的测定结果基本一致。
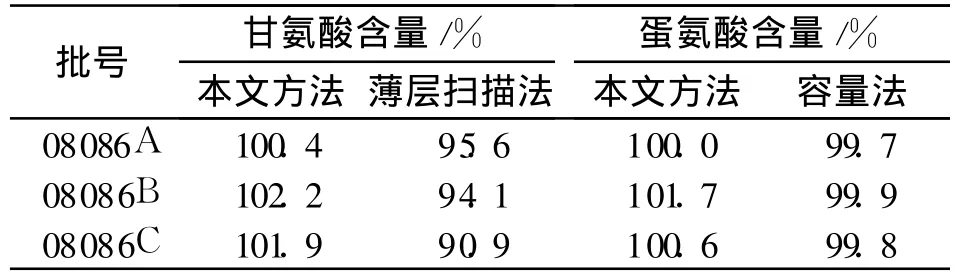
表2 不同方法测定甘氨酸与蛋氨酸含量的比较Tab.2 Determination results of glycine and methionine by different methods
3 讨 论
流动相的pH对分离度、峰形及保留时间有影响,pH为4.5时甘氨酸与相邻峰分离不好,蛋氨酸峰变形;pH为3.5时甘氨酸与相邻峰分离度好,但蛋氨酸峰分叉;pH为2.5时甘氨酸与蛋氨酸峰形均很好,理论板数均在10 000以上,甘氨酸与相邻峰的分离度大于2.0。流动相的pH对甘氨酸的保留时间基本无影响,随着 pH值减小,蛋氨酸的保留时间延长。
我们对检测波长进行了选择。甘氨酸和蛋氨酸均在200~220 nm波长范围内有吸收,属末端吸收,且吸光度随波长增大而减小;同时在200,203,205 nm进行检测,基线噪音基本相同,甘氨酸响应值减少明显,故选择 200 nm作为测定波长。
分别采用水和流动相为溶剂,色谱行为基本相同,故选用前者。由于检测波长为200 nm,对照品与供试品图谱中,甘氨酸峰后有一小峰。分别配制甘氨酸与蛋氨酸的对照品溶液,相应图谱中均有此峰;采用流动相配制,此峰亦存在;可能为水中少量的氯离子峰,向水中加入少量的盐酸溶液,进样,氯离子峰与上述小峰不完全对应;由于此小峰与甘氨酸分离度大于2.0,故不影响含量测定。另外,本品处方中还有甘草酸苷,由于本色谱系统中流动相洗脱能力较弱,甘草酸苷不出峰,故对本法无干扰。
本法与薄层扫描法、衍生化HPLC法比较,具有简便、快速、结果准确的优点。
[1]中国药品生物制品检定所.进口药品复核标准汇编[S].2000:373-374.
[2]田莉,高晓黎,云琦.反相高效液相色谱法测定复方甘草甜素片三组分的含量[J].新疆医科大学学报,2005,28(1):27-29.
[3]杜萍,孙明.HPLC法测定复方甘草酸苷胶囊中三组分的含量[J].中国药师,2007,10(10):1004-1006.
[4]张卫东.RP-HPLC法同时测定复方甘草酸苷片中三组分的含量[J].中国药物应用与检测,2009,6(3):153-155.
[5]靳颖华,张卫东,齐平.柱前衍生化HPLC法同时测定复方甘草酸苷胶囊中甘氨酸和蛋氨酸的含量[J].解放军药学学报,2009,25(2):172-174.
[6]吴小曼,纪宇.反相高效液相色谱法测定复方氨基酸注射液中的乙酰半胱氨酸和乙酰酪氨酸[J].中国生化药物杂志,2003,24(4):194-195.
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
中国饲料(2022年5期)2022-04-26
国外畜牧学·猪与禽(2018年8期)2018-05-14
国外畜牧学·猪与禽(2018年7期)2018-05-14
中国畜牧业(2016年10期)2016-02-21
人间(2015年11期)2016-01-09
中国畜牧业(2015年4期)2015-12-07
中国洗涤用品工业(2015年7期)2015-02-28
中国畜牧业(2014年4期)2014-10-16
天津药学(2013年2期)2013-12-23
