
大孔SiO2/ATO电极的制备及其电化学性能研究
2010-11-10 01:01:26李文丽张瑞丰王文钦
无机化学学报 2010年8期
李文丽 张瑞丰 王文钦 叶 剑
(宁波大学材料科学与化学工程学院,宁波 315211)
大孔SiO2/ATO电极的制备及其电化学性能研究
李文丽 张瑞丰*王文钦 叶 剑
(宁波大学材料科学与化学工程学院,宁波 315211)
以大孔SiO2为载体,通过SnCl2/SbCl3的乙二醇溶液的浸渍,孔道内两步水解和高温煅烧等处理,制备出大尺寸大孔径的ATO/SiO2导电材料,用SEM﹑FTIR﹑XRD﹑XPS对其进行结构表征,用稳态极化和苯胺电化学聚合研究其电化学特性。结果表明,ATO以10 nm尺寸的微粒形式均匀致密的负载在SiO2薄层上,电导率随ATO含量的增加而提高,负载三次后的体积电阻是18 Ω·cm,比表面积达到77 m2·g-1。ATO/SiO2大孔电极在酸性和中性条件下分别具有2.5和2.2 V的析氧电位。SiO2/ATO电极在恒电流下可以使苯胺发生电氧化聚合,并在ATO表面覆盖上聚苯胺导电膜,循环伏安实验表明聚苯胺具有电活性,并且电信号随着电极润湿程度的增加而增加,表明大孔电极的高比表面积对电化学反应有促进作用。
二氧化硅载体;导电氧化物;大孔电极;电化学性质
0 引 言
在现代电催化研究中,导电金属氧化物电极具有特殊的地位[1],被称为形稳阳极DSA(dimensionally stable anode)[2-4]。DSA电极不仅克服了铂电极费用高,石墨和铅基合金电极耐蚀性差﹑电催化性能低﹑电力消耗大等的缺点[5],而且还为电催化电极的制备提供了新思路。近年来研究发现,以Sb掺杂SnO2涂层的DSA电极,对有机物的氧化降解有很好的电催化作用,例如,以金属钛为支撑体的SnO2电极已经被国内外很多学者用做阳极,研究有机物的电氧化降解[6]。电催化氧化法用于处理难降解的有机物具有很好的效果,其独特优势体现在工艺简单,不需要复杂的专用设备,不需要使用其它的化学药剂,既降低成本,又不会引起二次污染。然而从实际应用的角度来说,现有的DSA电极仍然存在一些不足:涂层电极的电流效率低,对有机物的降解速率有待于进一步提高;电极稳定性差(电极表面活性层易脱落,电极使用寿命短[7]),因此围绕电极的材料科学问题,包括材料的组成﹑结构和制备方法等仍需要进一步的研究。
最近,我们制备了三维骨架结构的聚合物[8],并利用它作为整体型模板制备出大尺寸大孔径的SiO2载体[9]。这种新型SiO2具有三维超薄结构的特殊形貌,稳定性好,比表面积大(132 m2·g-1),孔隙率高(93%),非常适合制备含有功能材料的大孔复合材料。本论文以该材料为基础,结合溶胶/凝胶传统方法,开展了原位负载导电氧化物的研究,以SnCl2/SbCl3的乙二醇混合溶液为起始物,通过分步水解、高温煅烧等处理,生成掺杂锑的二氧化锡纳米微粒(Antimony-doped tin oxide,ATO), 利用 SiO2载体孔道内强大的毛细管效应,实现在化学转变过程中控制产物的形貌,使导电氧化物以纳米微粒的形式均匀附着在载体表面,获得导电性良好﹑耐氧化﹑耐腐蚀的ATO/SiO2大孔材料。这项工作的目的是使导电氧化物以一种新的结构存在于功能材料中,充分发挥其大孔骨架的优势,从而进一步改善ATO/SiO2电极的性能。
1 实验部分
1.1 SiO2载体的制备
基本步骤如下:首先取16.0 g环氧树脂在18.0 g聚乙二醇1000和14.0 g聚乙二醇2000混合,以4.0 g二乙烯三胺为固化剂,在70℃下固化反应3 h,水洗除去PEG后,便制得一种具有三维骨架结构的多孔聚合物。然后将上述聚合物模板在60℃下干燥2 h后,浸泡在正硅酸四乙酯中3 h,然后于50℃下放置在NH3·H2O的气氛中12 h使正硅酸四乙酯充分水解转化为SiO2,将得到复合物先在60℃下干燥2 h以除去反应生成的乙醇及吸附的NH3·H2O,然后置于马弗炉中以5℃·min-1的升温速率煅烧至820℃并维持30 min,去除聚合物模板后即得到与聚合物模板外形、尺寸一样的大孔SiO2,详细步骤可见文献[10]。
1.2 ATO/SiO2电极的制备及导电性的测量
将 15.21 g SnCl2·2H2O(67.42 mmol) 和 1.71 g SbCl3(7.496 mmol)溶解在24 g乙二醇中配成溶液,将上述SiO2载体浸泡到溶液中,随着气泡从载体中赶出,白色的载体逐渐变成透明,证明浸泡充分。将浸泡物取出后在90℃的烘箱中烘1 h,然后在氨气氛中放置2 h,完成第一阶段的原位水解。再将样品放入马弗炉中在3 h内逐渐从200℃升到400℃,此时溶剂完全蒸发。再将样品在稀氨水中浸泡40 min使氯化物原料完全水解,最后再放入马弗炉中从400℃升到700℃煅烧2 h,此时二价的锡完全被氧化成四价的锡形成ATO。上述步骤完成一次就是导电氧化物负载一次,多次重复就可成倍增加ATO的含量,ATO的负载量用称量的方法确定,即复合材料的总重量减去载体的重量。
导电性测量:由于该材料为多孔结构,它不同于一般的导电材料,若用一般的四探针法测量导电性,那么表面接触好坏可能会对导电性测量有影响,为了提高接触性能,我们在样品 (40 mm×15 mm×3 mm)的两端焊上一层锡再接出铜线,测量出其体积电阻。
1.3 ATO/SiO2电极的表征
采用JSM-5600LV型扫描电子显微镜 (SEM)观察ATO电极﹑PAN膜电极的形貌,即样品在5 kV的加速电压下进行表面形貌表征;采用PROTEGE 460 E.S.P,Nicolet型红外光谱仪(FTIR)对样品进行红外光谱分析,KBr压片;采用Rigaku D/max-1200型粉末X-射线衍射仪(XRD)在室温下测定样品的晶相,Cu Kα 辐射(λ=0.154 nm),电流为 30 mA,电压为40 kV,扫描范围 2θ=10°~55°; 采用岛津 Kratos AXIS-ULTRA DLD型多功能X射线光电子能谱(XPS)分析ATO电极的成份,射线源为Al Kα(Mono),150 W(10 mA,15 kV);采用 JW-K 型比表面积及孔径分布测试仪 (北京精微高博科学技术有限公司)对样品的比表面积进行分析,样品首先在真空下加热到150℃并维持4 h以去除样品吸附的物质,然后在77 K下进行测定,用BET公式计算其比表面积。
1.4 ATO/SiO2电极稳态极化曲线测量
电化学性能测试是在CHI660B电化学工作站上进行,测试温度为室温,采用三电极体系,以所制的ATO/SiO2电极为工作电极,Ti片为辅助电极,饱和甘汞电极为参比电极,分别在0.5 mol·L-1H2SO4溶液和 0.5 mol·L-1Na2SO4溶液中测量其伏-安特性(极化曲线)。
1.5 在ATO/SiO2电极上电化学合成聚苯胺
在电解槽中放入 0.2 mol·L-1的苯胺和 0.5 mol·L-1的硫酸溶液,以所制的ATO/SiO2电极为基体电极,聚合前通氮气30min以除去溶液中的氧气,在溶液静止的条件下,用恒电流法合成聚苯胺,用蒸馏水冲洗后马上放入0.5 mol·L-1的硫酸溶液中待测。用此电极作为工作电极,参比电极为222型饱和甘汞电极,辅助电极为铂丝电极,在除氧的0.5 mol·L-1的硫酸溶液中进行循环伏安测定。
2 结果与讨论
2.1 ATO电极的表征
2.1.1 形貌分析
图1中照片a是SiO2载体的SEM图片,由图可见SiO2薄层通过三维延伸构成宏观体,其中二氧化硅薄层的厚度约为27 nm,图1中照片b~d是ATO 含量分别为 53.7wt%(1次负载)﹑70wt%(2次负载)和77.4wt%(3次负载)的复合物的SEM结果,随着ATO含量的增加,基本骨架并未改变,薄层的形貌逐渐变得粗糙,说明导电氧化物是以纳米微粒方式均匀沉积在二氧化硅薄层表面。照片e是照片d的进一步放大,从中可见纳米微粒的致密排列,总体上来说纳米微粒的尺寸分布比较均匀,一般在10 nm左右,纳米ATO很可能是以由疏到密的方式原位生长,同时纳米微粒的尺寸会有一定程度的增大,尽管在制备过程中没有使用任何分散剂,但是从SEM照片中没有看到ATO颗粒的严重团聚现象,说明在微米级的孔道中制备纳米微粒要比自然状态下容易控制其形态。在孔道内的微环境有个特点,就是强大的毛细管效应对孔道内液体物质的作用力远大于液体本身的重力,这种情形等效于微重力状态,重力对形貌的影响远不如毛细管效应来的明显,毛细管效应的大小与表面的曲率有关,如果在第一次负载后某个位置出现空缺,那么在第二次负载时空缺的那个地方由于曲率大很可能被优先填平,这种自发修补的机制很可能是上述形貌容易控制的原因,也只有这样才能制备出导电性较好的复合材料。当然这种机制的存在也是有条件的,那就是溶胶/凝胶转变的过程必须足够的慢,我们使用高沸点的溶剂以及采取分步水解的办法正是出于这种考虑,如果转变的过程过快,那么ATO就会向样品的表面集中,而中心部分缺乏。照片f是ATO(77.4wt%)/SiO2在电氧化沉积聚苯胺(PAN)后的形貌,PAN完全覆盖了原来ATO的形貌,厚度明显增大,从电镜数据可以推测出复合层的厚度在50~70 nm之间,从整体上看PAN的沉积是均匀的,即样品的表面与中心部位之间没有明显的差异,说明在氧化聚合过程中聚合速率是比较均衡的。

图1 ATO/SiO2及SiO2载体的SEM图片Fig.1 SEM images of ATO/SiO2and SiO2support
2.1.2 红外光谱分析
ATO在二氧化硅上的负载可以通过红外光谱来反映,图2中曲线a是SiO2载体本身的红外光谱,800 cm-1为Si-O的对称伸缩振动吸收峰,1 100 cm-1为Si-O-Si的反对称伸缩振动吸收峰。3430和1 230 cm-1分别为薄层表面Si-O-H中O-H的伸缩振动和弯曲振动吸收峰,这是二氧化硅超薄层表面羟基大量存在的依据。曲线b~d是ATO含量分别为 53.7%,70%和 77.4%的 ATO/SiO2复合物红外光谱图。随着ATO含量的增加,3430 cm-1处和1230 cm-1处指示表面Si-O-H的峰越来越弱,说明ATO的负载改变了二氧化硅表面的状态,但是凭红外数据无法确定SiO2与ATO之间是否形成化学键。从红外光谱中还能看到ATO含量越大,其红外光的透过率越小,同时谱线的精细结构逐渐消失,这是由于ATO有导电性,其中的自由电子对红外光有较强的吸收。

图2 ATO/SiO2及SiO2的红外光谱图Fig.2 IR spectra of ATO/SiO2and SiO2。
2.1.3 XRD 分析
图 3是 ATO(77.4wt%)/SiO2的 XRD 结果,图中衍射峰的位置与四方形SnO2晶体结构的标准数据一致(No.88-2348),表明锑的掺杂没有改变二氧化锡的晶体结构和晶体参数[11]。Sb原子是取代(置换)或填隙(间隙)的方式进入SnO2晶格,因而在XRD中Sb没有以单独的氧化物形式存在[12]。SiO2载体是无定形的,没有衍射峰,只在2θ=23°左右出现较弱的非晶衍射包,与四方形金红石的SnO2(110)峰重叠。衍射峰的宽度反映了ATO颗粒的尺寸,根据谢乐公式(Scherrer):可以计算出图3中3个最强的布拉格衍射峰 (110)(101)(200)的晶粒半径分别是:D1=13.48 nm,D2=3.88 nm,D3=12.68 nm,平均晶粒半径D=10.02 nm,这个结果与电镜中观察到的纳米粒子尺寸相符。

图3 ATO含量为77.4%的ATO/SiO2大孔材料的XRD图Fig.3 XRD pattern of ATO/SiO2macroporous material(wATO=77.4%)
2.1.4 XPS 光电子能谱分析
图4是ATO(77.4wt%)/SiO2复合物的XPS测定结果,O1s的峰与Sb3d的峰发生了重叠,根据分峰结果O1s在530.6和 532.8 eV有2个峰,前者应为ATO中O1s的结合能,后者为SiO2中O1s的结合能。在ATO/SiO2复合物中,ATO的含量大约是SiO2的3倍,但是能谱显示SiO2中的氧原子数多于ATO中的氧原子,除了ATO的密度远大于SiO2的原因外,同时也说明ATO中有很多氧空位。Sb3d5/2的结合能为530.8 eV与O1s的结合能530.6 eV非常相近,Sb3d3/2的结合能为540.5 eV,由图可知Sb主要是以+5价形式存在。Si2p的结合能为103.6 eV,与能谱数据中的 Si2p 的结合能(103.2~103.7 eV)相符合。Sn3d5/2和 Sn3d3/2的结合能分别为 487.0和495.6 eV,峰差8.4 eV与Sn的标准能谱完全接近。估算Sn3d5/2和Sb3d3/2的峰面积,用到的灵敏度参数分别为 4.89(Sn3d5/2)和 4.80(Sb3d3/2),得出 Sb/Sn 原子数之比为1∶7.5,而在所用原料中Sb与Sn的数量比为1∶9,说明在负载过程的热处理阶段,Sn逸出的速率要高于Sb,很可能是氯化物的水解仍然不够充分,有待于进一步优化。

图4 ATO含量为77.4%的ATO/SiO2大孔材料的XPS谱Fig.4 XPS spectra of ATO/SiO2macroporous material(wATO=77.4%)
2.1.5 ATO含量对材料性能的影响
在SiO2载体上负载ATO是逐次进行的,少量多次的方式有利于ATO负载的均匀,但是效率太低。我们将SnCl2和SbCl3溶解在乙二醇中配成41.4wt%浓度的溶液作为原料,实验证明这个浓度是合适的,既保证了均匀性又提高了效率。表1是ATO负载次数的不同对ATO/SiO2复合物中ATO的含量、比表面积和ATO/SiO2体积电阻的影响。随着ATO负载次数的增加,ATO的含量也随之增加,同时ATO/SiO2复合物的导电性迅速提高。在负载过程中ATO微粒从分散到聚集,导电性随微粒相互接触程度的增大而提高。复合物的比表面积总体上呈下降趋势,但是ATO微粒本身对表面积有贡献,所以比表面积随ATO含量的增加下降的幅度不大。

表1 不同ATO含量的ATO/SiO2复合物的性质Table 1 Properties of ATO/SiO2composites with different content of ATO
2.2 ATO电极稳态极化曲线
在阳极上进行着有机物氧化降解和水分解析氧2个主要的竞争反应,因而要求电极具有较高的析氧超电势,从而有效抑制氧的析出(对于含Cl-比较多的废水,也可能是Cl2的析出)。图5a和b分别为常温下ATO/SiO2电极在0.5 mol·L-1H2SO4溶液和 0.5 mol·L-1Na2SO4溶液中的稳态极化曲线。无论在酸性还是在中性溶液中,ATO/SiO2电极都具较高的析氧电位。这一点与以金属钛为支撑体的ATO电极比较相似,高析氧电位使得阳极析氧的副反应不易发生而有利于羟基自由基的形成,且电流的效率相对较高[13]。
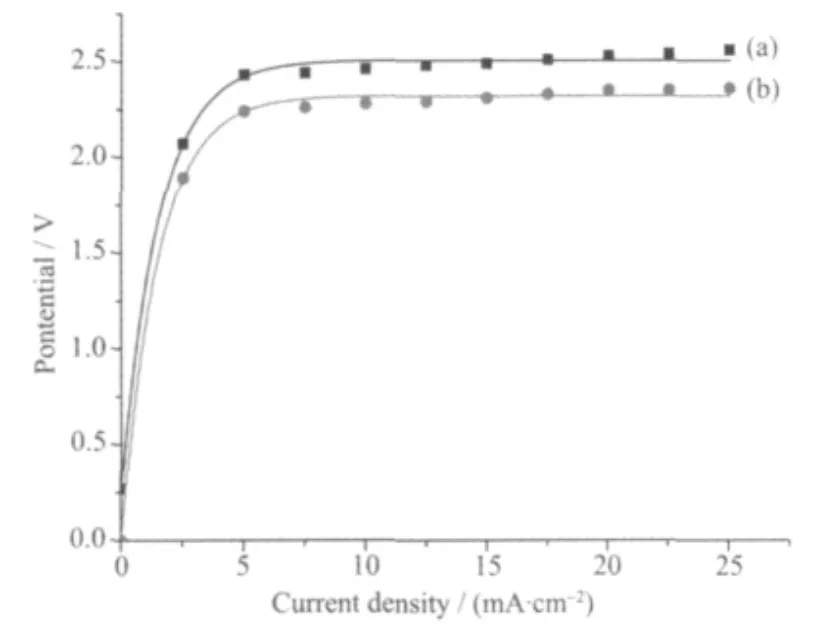
图5 ATO/SiO2电极在溶液中的稳态极化曲线Fig.5 Steady polarization curves of the ATO/SiO2 electrode in solution
2.3.1 沉积PAN的ATO/SiO2电极的循环伏安曲线
为了研究沉积聚苯胺的ATO/SiO2电极的电化学性质,控制电位在-0.4~1.0 V 之间,以 50 mV·s-1的扫描速度测得其在0.5 mol·L-1H2SO4溶液中的循环伏安曲线。图6中曲线a是干的电极刚浸泡在H2SO4溶液中所得到的CV曲线,可以看出其信号较弱。在H2SO4溶液中浸润5 min后,重新扫描得到曲线b,其中的氧化还原峰明显增大,再浸润10 min后扫描得到曲线c,聚苯胺的氧化还原峰进一步增高,基本达到最大值。这个实验说明,在浸润过程中电极的有效面积逐渐增大,发生电化学反应的场所增多,相应的信号也变大,这就是大孔电极所表现出的特性。在0~200 mV和600~800 mV 2个电位范围内分别存在2个可逆的氧化还原峰A和B。A被认为是聚苯胺从完全还原态(leuce-meraldine)到中间态(emeraldine)的氧化峰[14],在电位 200~300 mV和500~600 mV之间还有稍微小的氧化还原峰,这2个峰与聚合物降解产物密切相关[15]C1和C1′表示的是从苯亚胺氧化到苯醌状态,而C2和C2′是在稍高电位下氨基苯酚(p-aminophenol)和苯并喹啉(benzoquinonline)的氧化还原峰。纳米ATO电极呈现惰性,电极的析氧电位在2.2 V(对SCE)左右,此电极作为阳极电氧化合成反应电极,可避免析氧副反应的发生,测得的循环伏安图,与选用其他基底材料如Pt[16]﹑石墨棒[17]等电极沉积聚苯胺相同。
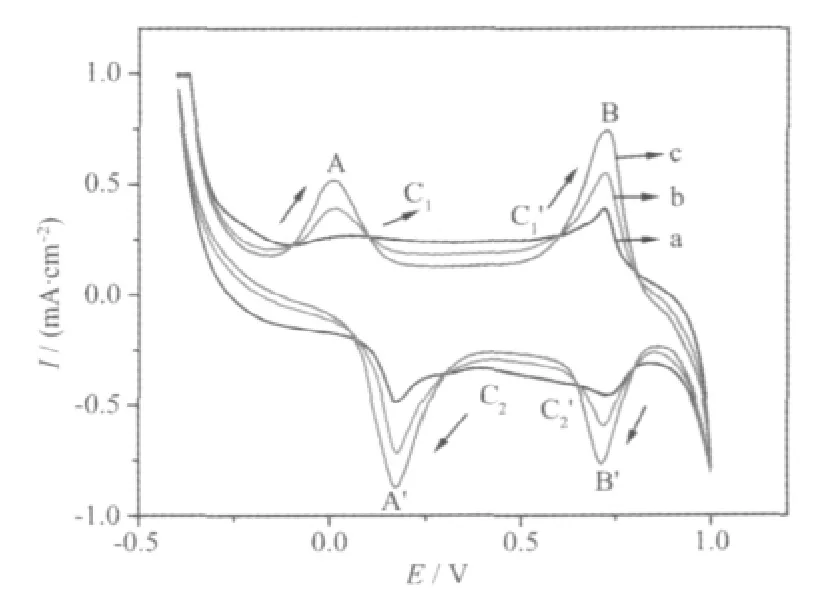
图6 恒电流合成聚苯胺的循环伏安曲线Fig.6 Cyclic voltammograms of polyaniline-deposited ATO/SiO2electrode
3 结 论
以大孔SiO2为载体,通过浸渍SnCl2/SbCl3的乙二醇混合溶液,孔道内分步水解和高温煅烧的方法制备出大尺寸大孔径的ATO/SiO2导电材料。经过多种手段表征证明导电氧化物ATO是以10 nm左右的微粒形式均匀负载在SiO2三维弯曲的薄层上。复合材料的导电性随ATO含量的增加而提高,经过3次负载体积电阻达到18 Ω·cm,比表面积达到77 m2·g-1。作为阳极的ATO/SiO2在酸性和中性介质中的析氧电位分别是2.5和2.2 V。用该电极可以电氧化沉积聚苯胺,沉积后电极的循环伏安曲线表现出浸润时间依赖性,即增大电极的有效面积对电化学反应有促进作用。
[1]Trasatti S.Electrodes of Conductive Metaltic Oxide.New York:Elsevier,1981.521-626
[2]Trasatti S.J.Electrochim.Acta,2000,45(15):2377-2385
[3]LIU Ye-Xiang(刘 业 翔 ).J.Central South Univ.Technol.(Zhongnan Gongye Daxue Xuebao),1996:99-102
[4]Lozano B C,Comninellis C,Battisti A D.J.Appl.Electrochem.,1997,27:99-974
[5]MENG Hui-Min(孟惠民),SHI Yan-Hua(史艳华),YUE Hong-Ying(俞宏英),et al.Mater.Protect.(Cailiao Baohu),2007,40(11):53-55
[6]Feng Y J,Li X Y.J.Water Res.,2003,37:2399-2407
[7]KONG De-Sheng(孔德生),LÜ Wen-Hua(吕文华),FENG Yuan-Yuan(冯媛媛),et al.Prog.Chem.(Huaxue Jinzhan),2009,21(6):1108-1117
[8]Zhang R F,Zhang L L.Polym.Bull.,2008,61:671-677
[9]LONG Neng-Bing(龙能兵),ZHANG Rui-Feng(张瑞丰).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(7):1153-1158
[10]Zhang R F,Long N B.J.Thin Solid Films,2009,517:6677-6680
[11]Zhang J R,Gao J.J.Mater.Lett.,2004,58(22/23):2730-2734
[12]Cui Y H,Feng Y J,Liu J F,et al.J.Funct.Mater.,2005,36(2):234-237
[13]Morita M,Iwakura C,Tamara H.J.Electrochim.Acta,1977,22(4):325-328
[14]Stilwell D E,Park S M.J.Electrochem.Soc.,1988,135(10):2254-2262
[15]Macdiarmid A G,Chiang J C,Richter A F.J.Synth.Met.,1987,18:285-290
[16]HOU Li-Bo(侯 丽 波),JIA Meng-Qiu(贾 梦 秋),HU Gang(胡刚),et al.J.Beijing Univ.Chem.Technol.(Beijing Huagong Daxue Xuebao),2004,31(4):65-69
[17]WANG Qin(王 琴),WU Ke-Zhong(武克忠),GUO Xiao-Li(郭小丽),et al.J.Hebei Normal Univ.(Hebei Shifan Daxue Xuebao),2007,31(2):204-207
Preparation and Electrochemical Properties of Macroporous ATO/SiO2Electrode
LI Wen-LiZHANG Rui-Feng*WANG Wen-Qin YE Jian
(Faculty of Material Science and Chemical Engineering,Ningbo University,Ningbo,Zhejiang 315211)
Macroporous ATO/SiO2conductive materials were prepared in large size by immersing macroporous SiO2supports into a mixed solution of SnCl2/SbCl3in ethylene glycol,a subsequent two-step in-situ hydrolysis and the final calcination at high temperature.The structure of the obtained material was characterized by means of SEM,FTIR,XRD and XRS.The electrochemical behavior was investigated by the steady polarization and electrochemical polymerization of aniline.The results showed that ATO formed nano-particles in an even size of 10 nm and packed closely on the SiO2thin layer.The conductivity increased with the content of ATO in the composite material,after three times of loading the volume resistance was measured to be 18 Ω·cm and the specific surface area was 77 m2·g-1.The macroporous ATO/SiO2anode showed an oxygen evolution potential in acidic and neutral conditons of 2.5 V and 2.2 V respectively.Aniline could be polymerized by the galvanostatic electropolymerization method and deposited on the ATO/SiO2electrode.Cyclic voltammetry experiments showed electrical activity of the deposited polyaniline,the electrical signal increased as the electrode was immersed in H2SO4for longer time,which indicated that a larger surface area provided a higher rate of electrochemical reaction on the macroporous electrode.
SiO2support;conductive oxide;macroporous electrode;electrochemical properties
O613.72;O611.2;O646.5
A
1001-4861(2010)08-1382-07
2010-03-01。收修改稿日期:2010-04-20。
国家自然科学基金(No.20674041),973前期专项(No.2010CB635116)及宁波大学王宽诚基金项目资助。*
。 E-mail:zhangruifeng@nbu.edu.cn
李文丽,女,26岁,硕士研究生;研究方向:功能材料。
猜你喜欢
中华环境(2021年9期)2021-10-14 07:51:06
中华环境(2021年8期)2021-10-13 07:28:34
中华环境(2021年7期)2021-08-14 01:57:26
陶瓷学报(2020年6期)2021-01-26 00:38:14
制造技术与机床(2018年10期)2018-10-13 06:36:56
疯狂英语·新悦读(2017年6期)2017-06-24 13:52:05
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
中国塑料(2015年7期)2015-10-14 01:02:34
中国塑料(2014年11期)2014-10-17 03:07:18
技术与教育(2014年2期)2014-04-18 09:21:33
