Pt-Se纳米空球修饰玻碳电极上甲酸的电催化氧化
2010-11-06 07:01颜亮亮江庆宁刘德宇温飞鹏邓小聪钟起玲田中群
物理化学学报 2010年9期
颜亮亮 江庆宁 刘德宇 钟 艳 温飞鹏 邓小聪钟起玲,* 任 斌,* 田中群
(1江西师范大学化学化工学院,南昌 330022; 2厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门 361005)
Pt-Se纳米空球修饰玻碳电极上甲酸的电催化氧化
颜亮亮1江庆宁2刘德宇2钟 艳1温飞鹏1邓小聪1钟起玲1,*任 斌2,*田中群2
(1江西师范大学化学化工学院,南昌 330022;2厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门 361005)
以无定形硒溶胶为模板制备了不同硒覆盖度(θSe)(θSe=0.49,0.39,0.06,0)的Pt-Se和Pt纳米空球(分别记为(Pt-Se)HN和PtHN),发展了利用亚硫酸盐彻底除去核壳纳米粒子上Se的方法.对获得的纳米空球进行了形貌和结构的表征,结果表明所制备的(Pt-Se)HN粒径均匀,分散性好,球壳呈多孔结构.以其作为电催化剂制备了(Pt-Se)HN修饰的玻碳(GC)电极((Pt-Se)HN/GC),利用常规电化学方法比较该电极与PtHN/GC和商用碳载铂(Pt/C)修饰GC (Pt/C/GC)电极对甲酸的催化氧化作用,发现对甲酸氧化的活性顺序为(Pt-Se)HN/GC>PtHN/GC>Pt/C/GC.三种电极催化甲酸氧化的机理有所不同:前者更倾向于通过弱吸附中间体直接氧化成CO2的单途径机理进行,后两者则通过强吸附和弱吸附中间体的双途径机理进行.在一定Se覆盖度条件下,(Pt-Se)HN/GC对甲酸的氧化有助催化作用.
电催化;Pt-Se纳米空球;(Pt-Se)HN/GC电极;甲酸;氧化
燃料电池因其高效、环境友好、安静、可靠性高等特点,被认为是21世纪首选的发电技术[1-2].甲酸是质子交换膜燃料电池的主要燃料之一,因其原料易得,毒性较小,分子结构相对简单等特点,近年来人们对甲酸的电催化氧化行为进行了较为广泛深入的研究[3-4].已有的研究表明,Pd虽然对甲酸有独特的催化活性,但因较易氧化而导致催化剂稳定性偏差[5-7];而Pt基催化剂尽管对甲酸氧化有较好的催化活性和稳定性,但纯Pt催化剂易被甲酸氧化所产生的强吸附中间体CO毒化而大大降低其催化活性[7-8].为了提高Pt基催化剂的活性及抗毒化能力,人们一方面试图利用小尺寸效应使其纳米化来提高其催化活性;另一方面在Pt上引入Ru、Au、Ag、Ge、Sn、Pb、As、Sb、Bi、Se、Te等物种组成二元或多元金属催化剂[9-12],利用所谓的“第三体效应”[13-15]来阻止CO在电极表面的吸附与毒化以提高其对甲酸等有机小分子氧化的催化活性.Llorca等[16]通过在Pt(111)上修饰不同量的Se发现,当Se覆盖度(θSe)大于0.20时,其催化甲酸的活性开始提高,θSe达到0.28时活性最大,但θSe超过0.33时,其催化活性消失.Motoo等[17-18]认为Se、Ag、Bi等作为一种非吸附氧物种修饰在Pt上对甲醇氧化起抑制作用,而对甲酸氧化起助催化作用.他们把Pt上修饰Se、Ag、Bi等元素对甲酸氧化催化活性的影响归结为反应所需最少活性位数目(SR)与邻位非活性原子数目(Shole)的相对变化所引起.Xia等[19]则认为Pb(不形成吸附氧)修饰在Pt上对甲酸氧化的助催化作用源于电子效应.
本文以硒溶胶为模板,合成了不同Se覆盖度(θSe=0.49,0.39,0.06,0)的Pt-Se和Pt纳米空球(分别记为(Pt-Se)HN和PtHN),将(Pt-Se)HN和PtHN分别组装到玻碳(GC)电极上,制得(Pt-Se)HN和PtHN修饰的玻碳((Pt-Se)HN/GC和PtHN/GC)电极.初步研究了不同Se覆盖度的(Pt-Se)HN/GC和PtHN/GC电极催化甲酸氧化的行为,并与商用碳载铂(Pt/C)修饰GC(Pt/C/GC)电极对甲酸的催化氧化性能进行了比较,为直接甲酸燃料电池阳极催化材料的研制积累了经验.
1 实 验
1.1 仪器与试剂
用日本日立公司生产的Hitachi S-4800扫描电镜(scanning electron microscopy,SEM)和荷兰FEI公司生产的TECNAI F30场发射高分辨透射电镜(high resolution transmission electron microscopy, HRTEM)观察所制备的纳米空球形貌;在TECNAI F30附带的EDAX公司生产的X射线能谱(energydispersive X-ray spectroscopy,EDX)仪上检测分析纳米空球组成;在荷兰PANalytical公司生产的X-pert PRO型X射线衍射仪上检测X射线衍射(X-ray diffraction,XRD)谱,管电流为30 mA,管电压为40 kV,Cu Kα为辐射源.RCT basic型控温磁力搅拌器由德国IKA WERKE公司生产,KQ5200型超声波清洗器由昆山市超声仪器有限公司生产.
电化学测量实验在CHI 631B型电化学工作站(上海辰华仪器有限公司生产)上进行.电解池是常规的三电极电解池:研究电极分别为(Pt-Se)HN/GC、PtHN/ GC和Pt/C/GC电极,其基底是直径为3 mm的GC电极 (从天津艾达恒晟科技发展有限公司购买);辅助电极为铂片电极;参比电极为饱和甘汞电极(SCE);所有电极电势都是相对于SCE.电化学研究体系均经通N2除氧,实验都是在25℃恒温水浴中进行.
商用碳载铂催化剂(40%(w)Pt/C)从E-TEK公司购买.实验中所用其它试剂均为分析纯.溶液用Millipore超纯水系统制备的电阻率≥18 MΩ·cm的超纯水配制.
1.2 (Pt-Se)HN,PtHN及其修饰GC电极的制备
将1 mL 0.1 mol·L-1的抗坏血酸溶液迅速加入至由十二烷基磺酸钠(SDSN)为保护剂的25 mL 1.0× 10-4mol·L-1的H2SeO3溶液中,在室温下搅拌30 min,制得砖红色的Se溶胶.接着将H2PtCl6溶液按Pt与Se的一定物质的量比滴加到Se溶胶中,同时加入过量的0.1 mol·L-1的抗坏血酸溶液,在室温下搅拌反应约5 h,即可得到硒核铂壳结构的实心纳米粒子(Se@Pt).将Se@Pt离心浓缩,加入水合肼静置若干小时,使核中的硒溶解在水合肼中并透过多孔的铂壳渗出[20],经多次离心洗涤后,就可得到铂壳上含有硒的Pt-Se纳米空球(Pt-Se)HN.若在100℃下,将Se@Pt实心纳米粒子与过量的亚硫酸钠在碱性条件下一起回流冷凝反应3 h,经离心清洗,亦可得到不含Se的Pt纳米空球PtHN.
分别将(Pt-Se)HN,PtHN,Pt/C与水及5%(w)Nafion按一定比例混合,超声15 min制得(Pt-Se)HN,PtHN和Pt/C的悬浮液,用移液枪移取5 μL悬浮液于GC电极上,在真空干燥器中真空干燥即可制得(Pt-Se)HN, PtHN和Pt/C修饰的GC((Pt-Se)HN/GC,PtHN/GC和Pt/ C/GC)电极.将(Pt-Se)HN/GC电极置于0.5 mol·L-1的硫酸溶液中,在-0.25-1.2 V电位区间进行循环伏安扫描以溶出铂壳中Se的方法来改变(Pt-Se)HN/GC电极中Se的覆盖度,用硒覆盖度不同的(Pt-Se)HN/GC电极研究催化甲酸氧化的活性.
2 结果与讨论
2.1 (Pt-Se)HN和PtHN的表征
图1为(Pt-Se)HN的SEM,TEM,HRTEM及选区电子衍射(selectedareaelectrondiffraction,SAED)图.从图1A可见,(Pt-Se)HN为球形,分散性好,粒径均匀,大小约90 nm;而图1B则证实(Pt-Se)HN为空心球形结构,球壳厚度约7 nm;从图1C可清楚地看到,(Pt-Se)HN球壳表面的晶格条纹,经测量计算为Pt(222)晶面;从图1D选区电子衍射图可知,(Pt-Se)HN球壳是多晶结构.有趣的是,(Pt-Se)HN球壳并不是完全密封的,而是由含有一定量Se的Pt原子团簇所构建的,具有很多纳米和亚纳米级孔洞的纳米壳,正是(Pt-Se)HN球壳上这些孔洞的存在为Se球模板的去除和电催化反应在球壳内表面的顺利进行提供了分子、离子等物种进出的通道;也正是这些孔洞的存在,成倍地增大了(Pt-Se)HN的比表面积和催化活性位数.多孔纳米空球球壳所具有的多级纳米结构,使其显示出优于其普通纳米粒子的金属性能.同样,在碱性介质中用Na2SO3代替水合肼除Se得到的PtHN形貌和结构与(Pt-Se)HN的基本相同.

图1 (Pt-Se)HN的扫描电镜(A),透射电镜(B),高分辨透射电镜(C)和选区电子衍射图(D)Fig.1SEM(A),TEM(B),HRTEM(C)images,and SAED pattern(D)of(Pt-Se)HN

图2 (Pt-Se)HN和PtHN的XRD谱图Fig.2 XRD patterns of(Pt-Se)HNand PtHN
图2 为 (Pt-Se)HN和PtHN的XRD谱.图2中衍射角位于39.9°、46.3°、67.8°、81.4°和85.8°左右的5个谱峰,分别对应于面心立方(fcc)铂的(111)、(200)、(220)、(311)和(222)晶面的特征衍射峰,但并未检则到Se晶体和铂的硒化物的特征衍射峰.比较(Pt-Se)HN与PtHN的XRD谱图可知,两者衍射谱峰几乎没有位移.图2的实验结果表明,Se在(Pt-Se)HN球壳中还是以无定形Se存在,且未影响到Pt的晶体结构.同时也印证了图1D中有关(Pt-Se)HN球壳结构是多晶Pt的事实.
图3为(Pt-Se)HN的EDX谱.从图3可以看到很强的Pt谱线和较弱的Se谱线,这表明将核壳结构的Se@Pt实心纳米粒子用水合肼浸泡清洗多次除核后所制备的Pt空球球壳上仍然有Se的存在.据此可以推测用水合肼浸泡清洗法难以将Se@Pt实心纳米粒子中的Se元素彻底除去.
2.2 (Pt-Se)HN/GC和PtHN/GC电极对甲酸的电催化氧化行为

图3 (Pt-Se)HN的EDX能谱图Fig.3 EDX spectrum of(Pt-Se)HN
图4为(Pt-Se)HN/GC,PtHN/GC和Pt/C/GC电极在0.5 mol·L-1H2SO4溶液中的循环伏安(CV)曲线.从图4可见,PtHN/GC与典型多晶铂电极在0.5 mol·L-1H2SO4溶液中的CV曲线基本相同;Pt/C/GC电极的CV曲线则稍有不同;而(Pt-Se)HN/GC电极的CV曲线则明显不同.在-0.24-0.1 V的电位区间,(Pt-Se)HN/ GC电极上由于Se的存在,使氢的吸、脱附受到很大的抑制.随后,在0.7-1.0 V的电位区间,出现了一相对较强的氧化峰.显然,该峰除了铂的氧化所作出的贡献外,其中还包含了Se的氧化[21]:

在Pt、Rh、Ir、Ni等金属表面会有H吸脱附,而目前尚没有文献报道H在Se上有吸脱附.在-0.24-0.20 V电位区间,H在单位面积Pt上的单层吸附或脱附所对应的电量为210 μC·cm-2,从而可以估算Pt的电化学活性面积[22].(Pt-Se)HN上Se的覆盖度(θSe)和Pt的电化学活性面积(AECSA)可分别由下述公式估算:

式中,θSe表示(Pt-Se)HN上Se的覆盖度,QH为H在PtHN上吸附或脱附所对应的电量,QSeH为H在(Pt-Se)HN上吸附或脱附所对应的电量.
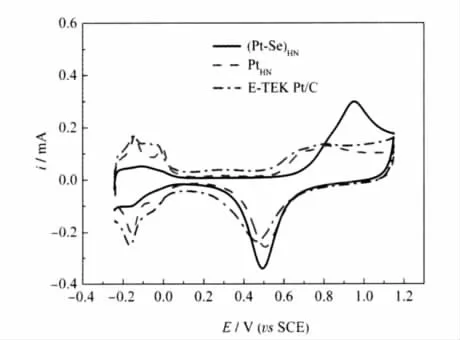
图4 (Pt-Se)HN/GC,PtHN/GC和Pt/C/GC电极在0.5 mol·L-1硫酸溶液中的循环伏安曲线Fig.4 Cyclic voltammetry of(Pt-Se)HN/GC,PtHN/GC, and Pt/C/GC electrodes in 0.5 mol·L-1H2SO4solutionscan rate:50 mV·s-1
图5 是(Pt-Se)HN/GC,PtHN/GC和Pt/C/GC电极在0.2 mol·L-1HCOOH+0.5 mol·L-1H2SO4溶液中的循环伏安曲线.PtHN/GC与Pt/C/GC电极催化甲酸氧化的CV曲线基本相似,正向扫描时,先后在约0.25和0.5 V左右出现两个氧化峰,这与多晶Pt电极催化甲酸氧化的CV曲线特征基本相同[23],可以用“双途径机理”[24-26]来解释.而(Pt-Se)HN/GC电极催化甲酸氧化的CV曲线与前两者明显不同,正向扫描时只在0.6 V左右有一个氧化峰,且其正扫与负扫两者的氧化电流几乎相同,所提供的单位面积总电量则比PtHN/GC和Pt/C/GC电极要大得多.显示出其催化甲酸氧化时产生的强吸附毒性中间体COad较少,催化活性较高的性质.人们普遍认为,甲酸氧化成CO需要两个相邻Pt的活性位,而甲酸直接氧化成CO2则不需要两个相邻Pt的活性位[27-28].可能由于(Pt-Se)HN/GC电极上Se的存在,一方面使得表面具有相邻Pt位的表面原子数目减少,因此强吸附毒性中间产物CO的数量也相应减少;另一方面当Pt纳米空球上修饰Se后,Se与Pt间的电子相互作用将可能对甲酸氧化有助催化作用[19],使(Pt-Se)HN/GC电极催化甲酸氧化可能更倾向于通过弱吸附中间体直接氧化成CO2的单途径机理进行.图5B为(Pt-Se)HN/GC,PtHN/GC和 Pt/C/GC电极在 0.2 mol·L-1HCOOH+0.5 mol·L-1H2SO4溶液中的计时电流曲线.从图5B可见,在电位阶跃至100 mV,催化甲酸氧化1800 s的过程中,(Pt-Se)HN/GC、PtHN/GC和Pt/C/ GC电极催化甲酸氧化的电流密度分别从400 s时的0.1、0.08和0.02 mA·cm-2降到0.06、0.03及0.01 mA·cm-2.比较上述三种电极催化甲酸氧化的计时电流曲线可知,(Pt-Se)HN/GC电极对甲酸催化氧化的活性最高,PtHN/GC次之,Pt/C/GC最低,这亦印证了图5A的实验结果.应该指出的是,PtHN/GC比Pt/C/ GC电极对甲酸催化氧化的活性更高,是因为PtHN的球壳与(Pt-Se)HN的球壳一样,并不是完全密封的,而是有很多纳米和亚纳米级孔洞.也正是这些孔洞的存在,成倍地增大了PtHN的比表面积和催化活性位数;亦为催化甲酸氧化反应在球壳内表面的顺利进行提供了分子、离子等物种进出的通道.

图5 甲酸在(Pt-Se)HN/GC,PtHN/GC和Pt/C/GC电极上的循环伏安(A)与计时电流(B)曲线Fig.5 Cyclic voltammetry(A)and chronoamperometric(B)curves of formic acid on(Pt-Se)HN/GC, PtHN/GC,and Pt/C/GC electrodessolution:0.2 mol·L-1HCOOH+0.5 mol·L-1H2SO4;scan rate:50 mV·s-1;potential stepped from-0.2 to 0.1 V(vs SCE)

图6 甲酸在不同硒覆盖度的(Pt-Se)HN/GC电极上的循环伏安(A)与计时电流(B)曲线Fig.6 Cyclic voltammetry(A)and chronoamperometric(B)curves of formic acid on(Pt-Se)HN/GC electrode with different coverages of Sesolution:0.1 mol·L-1HCOOH+0.5 mol·L-1H2SO4;scan rate:50 mV·s-1;potential stepped from-0.2 to 0.1 V(vs SCE)
图6 为不同硒覆盖度(θSe=0.49,0.39,0.06,0)的(Pt-Se)HN/GC电极在0.1 mol·L-1HCOOH+0.5 mol· L-1H2SO4溶液中催化甲酸氧化的循环伏安和计时电流曲线.从图6可见,当θSe=0.39时,(Pt-Se)HN/GC电极对甲酸的电催化氧化活性达到最高,但当Se覆盖度过高(θSe=0.49)时,其对甲酸的电催化氧化活性反而降低.图6的实验结果表明,在一定的Se覆盖度范围内,Se覆盖度的增加,有利于提高(Pt-Se)HN/ GC电极对甲酸的电催化氧化活性.这种随Se覆盖度的增加,铂对甲酸氧化的催化活性先升后降的“火山型”关系与Llorca等[16]的报道结果相似,只是由于计算θSe的方法不同,所制备与使用的铂催化材料的结构不同,Pt-Se催化剂的组成在数值上亦有所不同.
3 结 论
本文以无定形硒溶胶为模板合成了核壳结构的Se@Pt纳米粒子,用化学除核与电化学除硒方法相结合,制备了不同硒覆盖度(θSe=0.49,0.39,0.06,0)的(Pt-Se)HN和PtHN,并开展了相关的表征和甲酸电催化氧化研究,得出以下几点主要结论:
(1)用亚硫酸盐能够将Se@Pt核壳结构实心纳米粒子中的Se元素彻底除去,而用水合肼浸泡清洗法则难以将其中的Se元素彻底除去.所制备的(Pt-Se)HN粒径均匀,分散性好,球壳呈多孔结构,其中的残留Se还是以无定形Se的形式存在.
(2)(Pt-Se)HN/GC电极中的Se,在一定的范围内对甲酸氧化有助催化作用.
(3)(Pt-Se)HN/GC、PtHN/GC和Pt/C/GC电极催化甲酸氧化的机理有所不同,前者催化甲酸氧化更倾向于通过弱吸附中间体直接氧化成CO2的单途径机理进行;后两者则通过强吸附和弱吸附中间体双途径机理进行.它们对甲酸氧化的活性大小顺序为(Pt-Se)HN/GC>PtHN/GC>Pt/C/GC.
1 Mao,Z.Q.Fuel cell.Beijing:Chemical Industry Press,2005:1-31 [毛宗强.燃料电池.北京:化学工业出版社,2005:1-31]
2 Yi,B.L.Fuel cell:foundmental,technology,application.Beijing: Chemical Industry Press,2003:1-7 [衣宝廉.燃料电池:原理·技术·应用.北京:化学工业出版社,2003:1-7]
3 Lu,T.H.High-Technology and Industrialization,2008,11:70 [陆天虹.高技术与工业,2008,11:70]
4 Hong,P.;Liao,S.J.Modern Chemical Industry,2009,8:15[洪 平,廖世军.现代化工,2009,8:15]
5 Ha,S.;Dunbar,Z.;Masel,R.I.J.Power Sources,2006,158:129
6 Yang,G.X.;Chen,T.T.;Tang,Y.W.;Lu,T.H.Acta Phys.-Chim. Sin.,2009,25(12):2450 [杨改秀,陈婷婷,唐亚文,陆天虹.物理化学学报,2009,25(12):2450]
7 Yu,X.W.;Pickup,P.G.J.Power Sources,2008,182:124
8 Zhang,S.S.;Yuan,X.Z.;Hin,J.N.C.;Wang,H.J.;Friedrich,K. A.;Schulze,M.J.Power Sources,2009,194:588
9 Motoo,S.Proceedings of the Symposium chemistry and physics of electrocatalysis.McIntyre,J.D.E.;Weaver,M.J.;Yeager,E.B.Eds. SanFrancisco,1983,Princeton,NJ:The Electrochemical Society, 1984:331
10 Rice,C.;Ha,S.;Masel,R.I.;Waszczuk,P.;Wieckowski,A.; Barnard,T.J.Power Sources,2002,111:83
11 Rice,C.;Ha,S.;Masel,R.I.;Wieckowski,A.J.Power Sources, 2003,115:229
12 Uhm,S.;Lee,H.J.;Kwon,Y.;Lee,J.Angew.Chem.Int.Edit., 2008,47:10163
13 Angerstein-Kozlowska,H.;MacDougall,B.;Conway,B.E. J.Electrochem.Soc.,1973,120:756
14 Adzic,R.R.;Simic,D.N.;Despic,A.R.;Drazic,D.M. J.Electroanal.Chem.,1975,65:587
15 Zhang,H.X.;Chao,W.;Wang,J.Y.;Zhai,J.J.;Cai,W.B. J.Phys.Chem.C,2010,114:6446
16 Llorca,M.J.;Herrero,E.;Feliu,J.M.;Aldaz,A.J.Electroanal. Chem.,1994,373:217
17 Shibata,M.;Motoo,S.J.Electroanal.Chem.,1987,229:385
18 Shibata,M.;Takahashi,O.;Motoo,S.J.Electroanal.Chem.,1988, 249:253
19 Xia,X.H.;Iwasita,T.J.Electrochem.Soc.,1993,140:2559
20 Chizhikov,D.M.;Shchastlivyi,V.P.Selenium and selenides (translated from the Russion by Elkin,E.M.).London and Wellingborough:Collet′s LTD,1968:40
21 Wang,R.F.;Liao,S.J.;Liu,H.Y.;Meng,H.J.Power Sources, 2007,171:471
22 Trasatti,S.;Petrii,O.A.Pure Appl.Chem.,1991,63:711
23 Wu,Y.N.;Liao,S.J.Acta Phys.-Chim.Sin.,2010,26(3):669 [吴燕妮,廖世军.物理化学学报,2010,26(3):669]
24 Leiva,E.;Iwasita,T.;Herrero,E.;Feliu,J.M.Langmuir,1997, 13:6287
25 Samjeské,G.;Osawa,M.Angew.Chem.Int.Edit.,2005,44:5694
26 Park,I.S.;Lee,K.S.;Choi,J.H.;Park,H.Y.;Sung,Y.E.J.Phys. Chem.C,2007,111:19126
27 Park,S.;Xie,Y.;Weaver,M.J.Langmuir,2002,18:5792
28 Kristian,N.;Yan,Y.;Wang,X.Chem.Commun.,2008:353
Electrocatalytic Oxidation of Formic Acid on Pt-Se Hollow Nanosphere Modified Glassy Carbon Electrodes
YAN Liang-Liang1JIANG Qing-Ning2LIU De-Yu2ZHONG Yan1WEN Fei-Peng1DENG Xiao-Cong1ZHONG Qi-Ling1,*REN Bin2,*TIAN Zhong-Qun2
(1College of Chemistry and Chemical Engineering,Jiangxi Normal University,Nanchang 330022,P.R.China;2State Key Laboratory of Physical Chemistry of Solid Surfaces,Department of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,Fujian Province,P.R.China)
Platinum-selenium and platinum hollow nanospheres(denoted as(Pt-Se)HNand PtHN,respectively)with differentcoveragesofSe(θSe)(θSe=0.49,0.39,0.06,0)were prepared using amorphous Se colloids as a sacrificial template. Sulfite was used to completely remove Se from the core-shell nanoparticles.The morphology and structure of the nanoparticles were characterized using various methods,which revealed a hollow structure with a very uniform size distributionandaporousstructureontheshell.AssemblyofPt-Sehollownanospheres((Pt-Se)HN)on a glassy carbon(GC) electrode produced a(Pt-Se)HN/GC electrode.The electrocatalytic activity of the electrode for the oxidation of formic acidwascomparedwiththePtHN/GCandcommercialPt/C/GCelectrodesbycyclicvoltammetryand chronoamperometry. The activity followed the order:(Pt-Se)HN/GC>PtHN/GC>Pt/C/GC.The electrooxidation of formic acid on(Pt-Se)HN/C, PtHN/C,and Pt/C catalysts follows different mechanisms:the former tends to directly oxidize formic acid to CO2via weakly adsorbed intermediates,and the latter two via both weakly and strongly adsorbed intermediates.(Pt-Se)HNwith a suitable selenium content showed optimal electrocatalytic activity for the oxidation of formic acid.
Electrocatalysis;Pt-Se hollow nanosphere;(Pt-Se)HN/GC electrode;Formic acid; Oxidation
O646
Received:March 11,2010;Revised:May 14,2010;Published on Web:June 30,2010.
*Corresponding authors.Email:zhqiling@163.com,bren@xmu.edu.cn;Tel:+86-13879159319.
The project was supported by the National Natural Science Foundation of China(20663002),Foundation of State Key Laboratory of Physical Chemistry of Solid Surfaces,Xiamen University,China(200511).
国家自然科学基金(20663002)和厦门大学固体表面物理化学国家重点实验室基金(200511)资助项目
ⒸEditorial office of Acta Physico-Chimica Sinica
猜你喜欢
科学技术创新(2022年30期)2022-10-21
农业与技术(2021年23期)2021-12-14
石油化工设备(2021年6期)2021-11-20
黑龙江水利科技(2020年8期)2021-01-21
科学技术创新(2020年27期)2020-09-05
原子与分子物理学报(2020年5期)2020-03-17
中国有色金属学报(2018年2期)2018-03-26
河北建筑工程学院学报(2017年3期)2017-12-15
中南大学学报(自然科学版)(2016年2期)2017-01-19
光学精密工程(2016年5期)2016-11-07