三重态类硅烯HB═SiLiF的结构及其与R—H (R=F,OH,NH2)的插入反应
2010-11-06 07:01李文佐祝洪杰程建波李庆忠宫宝安
物理化学学报 2010年9期
李文佐 祝洪杰 程建波 李庆忠 宫宝安
(烟台大学化学生物理工学院,山东烟台 264005)
三重态类硅烯HB═SiLiF的结构及其与R—H (R=F,OH,NH2)的插入反应
李文佐*祝洪杰 程建波 李庆忠 宫宝安
(烟台大学化学生物理工学院,山东烟台 264005)
用密度泛函理论(DFT)和二次组态相互作用(QCISD)方法研究了三重态类硅烯HB═SiLiF的结构及其与RH(R=F,OH,NH2)的插入反应.计算结果表明,类硅烯HB═SiLiF有三种平衡构型,其中四元环构型能量最低,是其存在的主要构型.HB═SiLiF与HF,H2O和NH3发生插入反应的机理相同.QCISD/6-311++G(d,p)// B3LYP/6-311+G(d,p)计算的三个反应的势垒分别为124.85,140.67和148.16 kJ·mol-1,反应热分别为-2.22,20.08和23.22 kJ·mol-1.相同条件下发生插入反应时,反应活性都是H—F>H—OH>H—NH2.
插入反应;类硅烯HB═SiLiF;B3LYP;QCISD
类硅烯(silylenoid)是硅烯(silylene)的一种衍生物,通常可以表示为R1R2SiMX(M为碱金属原子, X为卤素原子).一般来说类硅烯比其相对应的硅烯稳定,在有机合成中类硅烯是一种重要的有机硅活性中间体.最近二十多年,人们对类硅烯进行了大量研究.1995年,Tamao和Kawachi[1]首次对类硅烯进行了实验研究,他们证实了类硅烯Ph2SiLi(OBu-t) ([(tert-butoxy)diphenylsilyl]lithium)的存在并研究了其性质.2004年,Lee等[2]在室温下合成了稳定的类硅烯(Tsi)X2SiLi(Tsi=C(SiMe3)3;X=Br,Cl).2006年,Molev等[3]报道了第一个成功分离出的含F类硅烯(R3Si)2SiFLi·3THF(R3Si=t-Bu2MeSi)的分子结构.其它一些实验[4-12]也证实类硅烯是重要的活性中间体.对类硅烯的理论研究也很多.自从Clark等[13]首次用从头算方法研究了最简单的类硅烯H2SiLiF的结构,至今人们已较系统地研究了多种类硅烯的结构、稳定性、异构化反应、插入反应、加成反应、取代反应和消去反应等[14-37].
与卡宾不同,大部分硅烯的基态是单重态[38-40].因此,以往所研究的类硅烯也均是单重态.然而,人们也发现有些硅烯的基态是三重态[41-47].那么,基态为三重态的类硅烯是否也存在?如果存在,其结构、稳定性与反应性如何?本文中我们通过理论计算,发现了一些基态为三重态的类硅烯,HB═SiLiF是其中之一.插入反应是类硅烯较常见的反应类型[36-37],本文研究了HB═SiLiF的结构及其与RH(R=F, OH,NH2)的插入反应.
1 计算方法
本工作所用研究方法与我们以往对类硅烯的研究所用方法相同[30-31,35,37].利用密度泛函理论(DFT) B3LYP[48-49]方法优化所有驻点的构型,计算时选择6-311+G(d,p)基组[50].对优化所得的每一个构型在相同计算水平上进行振动频率分析计算以确定驻点性质.对涉及的过渡态构型,在同一计算水平上进行内禀反应坐标(IRC)[51]计算.为了进一步考虑相关能,在优化构型上对各个物种进行了 QCISD[52-53]/6-311++G(d,p)单点能量计算.如无特殊说明,文中能量均指QCISD/6-311++G(d,p)//B3LYP/6-311+G(d,p)水平上计算所得的能量,并在B3LYP/6-311+G(d,p)水平上进行零点能校正.所有计算均采用Gaussian 03[54]程序.
2 结果和讨论
众所周知,硅烯H2Si:的基态是单重态[55].然而,计算发现硅烯HB═Si:的基态是三重态.硅烯HB═Si:的示意图见图1.从表1可以看出,三重态的HB═Si:的能量比单重态的HB═Si:的能量低58.87 kJ·mol-1,因此三重态的HB═Si:比单重态的HB═Si:稳定.三重态类硅烯HB═SiLiF可看作三重态的HB═Si:与LiF结合而成.事实上,我们也计算了单重态的类硅烯HB═SiLiF,结果发现HB═SiLiF的单重态的能量比三重态的高.在QCISD/6-311++G (d,p)//B3LYP/6-311+G(d,p)水平上,单重态比三重态的能量高9.46 kJ·mol-1.因此本文只报道三重态的HB═SiLiF的相关结果,下文出现的HB═SiLiF均指三重态.B3LYP/6-311+G(d,p)优化的HB═SiLiF的可能构型在图2给出.各构型的总能量、零点振动能和相对能量列于表2.

图1 硅烯HB═Si:的示意图Fig.1 Schematic diagram of silylene HB═Si: bond length in nm
2.1 HB═SiLiF的平衡构型
B3LYP/6-311+G(d,p)计算表明三重态类硅烯HB═SiLiF有三种平衡构型(图2中1-3).如图2所示,构型1是四元环结构.构型1中所有原子在同一平面内,该构型属于Cs点群.构型1中的Li—F键比LiF中的长0.0176 nm,B—Si键比HB═Si:中的长0.0077 nm,说明构型1中的Li—F键和B—Si键比LiF和HB═Si:中的键弱.在三个构型中构型1能量最低.构型1的能量比HB═Si:和LiF的能量之和低133.68 kJ·mol-1.构型2是三元环结构,也属于Cs点群.构型2可看作LiF的Li端和F端分别接近Si的sp和p轨道形成Si—Li和F—Si授受键从而形成的配合物.构型2中Li—F键比LiF中的长0.0131 nm,B—Si键则比HB═Si:中的短0.0016 nm,说明构型2中的Li—F键比LiF中的弱,而B—Si键比HB═Si:中的强.构型2的能量比HB═Si:和LiF的能量和低65.44 kJ·mol-1.从表2可以看出,构型2的能量比构型1的能量高68.24 kJ·mol-1.从图2可以看出,构型3几乎是一个线型结构.构型3中Li—F键比LiF中的长0.0014 nm,B—Si键则比HB═Si:中的短0.0017 nm,说明构型3中的Li—F键比LiF中的弱,而B—Si键比HB═Si:中的强.构型3的能量比HB═Si:和LiF的能量和低42.13 kJ·mol-1,比构型1的能量高91.55 kJ·mol-1(见表2).
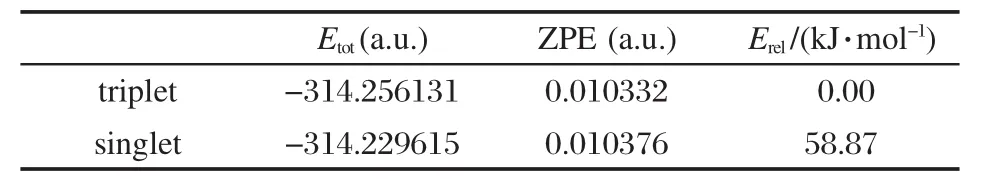
表1 硅烯HB═Si:的三重态和单重态的总能量(Etot)、零点振动能(ZPE)和相对能量(Erel)Table 1 Total energies(Etot),zero-point vibrational energies(ZPE),and relative energies(Erel)of the triplet and singlet silylene HB═Si:

图2 B3LYP/6-311+G(d,p)水平上优化的HB═SiLiF构型Fig.2 Optimized configurations of HB═SiLiF calculated at the B3LYP/6-311+G(d,p)levelbond length in nm and angle in degree
总之,类硅烯HB═SiLiF有三种平衡构型,从各自的能量比较各构型的稳定性顺序应为1>2>3.
2.2 HB═SiLiF与R—H(R=F,OH,NH2)的插入反应
类硅烯HB═SiLiF与R—H(R=F,OH,NH2)的插入反应按下式进行:
HB═SiLiF+R—H→HB═SiRH+LiF
如前所述,三重态类硅烯HB═SiLiF的四元环结构(即构型1)能量最低,应该是其存在的主要构型,因此在研究HB═SiLiF与R—H(R=F,OH,NH2)的插入反应时把构型1作为反应物.下文所提及的HB═SiLiF均指构型1.反应仅考虑三重态势能面.计算发现,发生插入反应时,先经历一个前驱体复合物(用Q表示),然后经历一个过渡态(TS)和一个中间体(IM)到达产物(P).B3LYP/6-311+G(d,p)构型优化所得各个驻点的几何构型如图3所示.各个驻点的相对能量列于表3.
2.2.1 前驱体复合物
从图3可以看出,三个前驱体复合物Q1、Q2和Q3有类似的结构.前驱体复合物中的“HB═SiLiF”部分与构型1非常接近.Si,H2和X(X=F,O,N)原子几乎在一条线上,即X=F时,θ(SiH2F2)=179.4°;当X=O时,θ(SiH2O)=179.2°;当X=N时,θ(SiH2N)= 179.4°.从表3可以看出,每一个插入反应的前驱体复合物的能量比反应物(HB═SiLiF+R—H)的总能量低,即X=F时,低12.68 kJ·mol-1;X=O时,低6.36 kJ·mol-1;X=N时,低2.22 kJ·mol-1.

表2 类硅烯HB═SiLiF各构型的总能量(Etot)、零点振动能(ZPE)和相对能量(Erel)Table 2 Total energies(Etot),vibrational zero-point energies(ZPE),and relative energies(Erel)of the configurations of the triplet silylenoid HB═SiLiF
2.2.2 过渡态
从图3可以看出,三个过渡态TS1、TS2和TS3也有类似的结构.每个过渡态结构中都有一个三元环X—Si—H2(X=F,O,N).各过渡态中的Si—F键均比HB═SiLiF中的Si—F键略短,即R=F时,短0.0066 nm;R=O时,短0.0034 nm;R=N时,短0.0036 nm.三个过渡态中的R—H2键键长均比反应物RH中的长,即TS1中,F2—H2键(0.1275 nm)比HF中F—H键(0.0922 nm)长0.0353 nm;TS2中,O—H2键(0.1304 nm)比H2O中的O—H键(0.0962 nm)长0.0342 nm;TS3中,N—H2键(0.1405 nm)比NH3中N—H键(0.1015 nm)长0.0390 nm.R—H键键长的明显增大意味着该键将要断裂,同时新的Si—H2键和Si—X键将要形成.
频率分析计算表明三个过渡态均存在唯一的虚频.在B3LYP/6-311+G(d,p)水平上,TS1,TS2和TS3的虚频分别为1297.5i,1490.5i和1528.8i cm-1.IRC计算表明,过渡态连接前驱体复合物与中间体.过渡态TS1,TS2和TS3相对于各自前驱体复合物(Q)的相对能量分别为124.85,140.67和148.16 kJ·mol-1(见表3).
2.2.3 中间体和产物
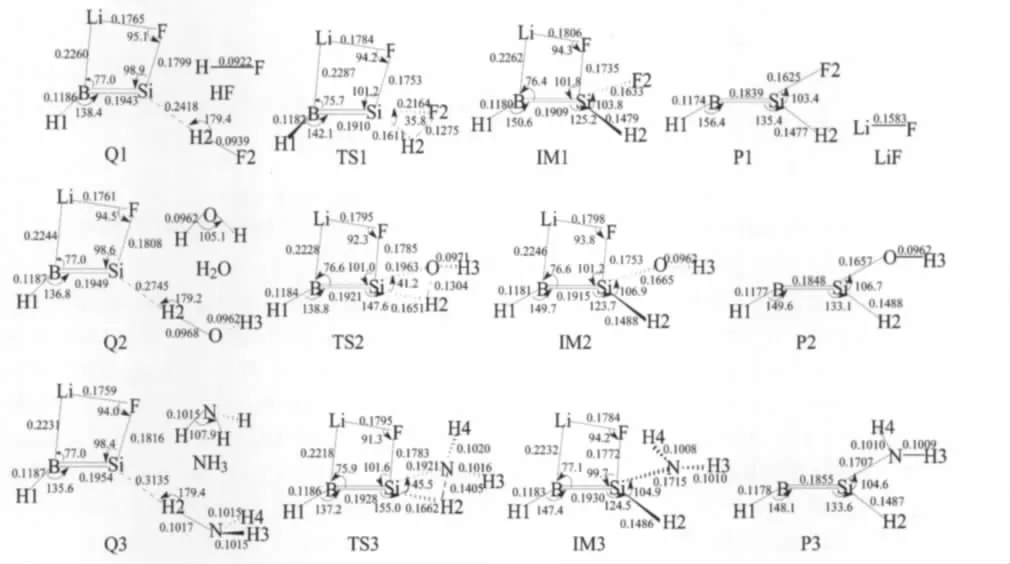
图3 B3LYP/6-311+G(d,p)水平上优化的反应HB═SiLiF+R—H各驻点的构型Fig.3 Geometries of the stationary points in the reaction HB═SiLiF+R—H calculated at the B3LYP/6-311+G(d,p)levelbond length in nm and angle in degree
IM1,IM2和IM3分别是三个插入反应的中间体(见图3).很明显,三个中间体也具有类似的结构.比较中间体与对应过渡态的结构,可以看出,中间体的θ(XSiH2)(X=F,O,N)键角明显大于过渡态的θ(XSiH2)键角,即IM1的θ(F2SiH2)(103.8°)比TS1的键角θ(F2SiH2)(35.8°)大68.0°;IM2的θ(OSiH2) (106.9°)比TS2的θ(OSiH2)(41.2°)大65.7°;IM3的θ(NSiH2)(104.9°)比TS3的θ(NSiH2)(45.5°)大59.4°.中间体构型中,R—H2键已完全断裂.三个中间体中的Si—H2键和Si—X(X=F,O,N)键均比各自对应的过渡态中的Si—H2键和Si—X键短,即IM1中的Si—H2键(0.1479 nm)比TS1中Si—H2键(0.1611 nm)短0.0132 nm,Si—F2键(0.1633 nm)比TS1中Si—F2键(0.2164 nm)短0.0531 nm;IM2中的Si—H2键(0.1488 nm)比TS2中Si—H2键(0.1651 nm)短0.0163 nm,Si—O键(0.1665 nm)比TS2中的Si—O键(0.1963 nm)短0.0298 nm;IM3中Si—H2键(0.1486 nm)比 TS3中 Si—H2键(0.1662 nm)短0.0176 nm,Si—N键(0.1715 nm)比TS3中Si—N键(0.1921 nm)短0.0206 nm.说明新的Si—H2和Si—X键已经基本形成.从表3可以看出,反应过程的各个驻点中中间体的能量最低.IM1,IM2和IM3相对于各自前驱体复合物的相对能量分别为-205.56,-177.69和-157.78 kJ·mol-1(见表3).
P1,P2和P3(见图3)分别是三个中间体IM1, IM2和IM3解离掉LiF后的产物.计算表明,解离过程为能量单调升高的无势垒过程.P1,P2和P3均属于Cs点群.从表3可以看出,三个插入反应的产物(HB═SiRH+LiF)的能量均比各自的前驱体复合物的能量高,R=F,OH,NH2时,产物能量分别比复合物能量高10.46、26.44和25.44 kJ·mol-1.

表3 反应HB═SiLiF+R—H的反应物、过渡态、中间体和产物的相对能量(kJ·mol-1)Table 3 Relative energies(in kJ·mol-1)of reactants, transition states,intermediates,and products in the reaction HB═SiLiF+R—H
2.2.4 插入反应的机理分析

图4 HB═SiLiF与HF反应的能量和键长随反应坐标的变化曲线Fig.4 Potential energy and bond distance profiles along the reaction coordinates for the reaction of HB═SiLiF and HF
以HB═SiLiF与HF反应为例.以计算的TS1为起始点,在B3LYP/6-311+G(d,p)水平上沿着反应途径分别向前和向后进行了20个点的IRC计算,所用步长为0.15 a.u..图4给出了IRC计算所得的能量及键长随反应坐标变化的曲线图.从图4可以很直观地看出,随着插入反应的进行,F2—H2键逐渐拉长乃至断裂.Si—F2和Si—H2键逐渐缩短.
反应过程各个驻点的原子上的电荷变化可以反映反应机理.在B3LYP/6-311+G(d,p)水平上计算了各个前驱体复合物、过渡态、中间体和产物的Mulliken电荷.结果发现,过渡态中的X(X=F,O,N)原子和H2原子上的负电荷都比它们在复合物中的更多,Si原子上的正电荷则比在复合物中的更多.中间体中的X(X=F,O,N)原子和H2原子上的负电荷都比它们在过渡态中的更多.这同样可以反映出反应过程中X—H键的断裂和Si—H2与Si—X键的生成.
2.2.5 三个插入反应的比较
虽然HB═SiLiF与R—H(R=F,OH,NH2)发生插入反应的机理相同,但反应的难易程度不同. QCISD/6-311++G(d,p)//B3LYP/6-311+G(d,p)水平计算的HB═SiLiF与HF,H2O和NH3插入反应的势垒分别为124.85、140.67和148.16 kJ·mol-1(见表3),反应热分别为-2.22、20.08和23.22 kJ·mol-1.因此,不管是从热力学角度还是从动力学角度看,在相同条件下发生插入反应时,反应活性顺序都是H—F>H—OH>H—NH2,即相同条件下,HF最容易与HB═SiLiF发生插入反应,H2O次之,NH3则最难.这个顺序与RH中H原子上的电荷分布情况相一致:B3LYP/6-311+G(d,p)水平计算的HF,H2O和NH3的 H原子上的Mulliken电荷分别为 0.286e、0.253e和0.227e,当发生类似的亲核反应时,反应活性顺序应该是H—F>H—OH>H—NH2.HB═SiLiF与R—H(R=F,OH,NH2)的插入反应与其它类硅烯[37]与R—H(R=F,OH,NH2)发生插入反应的情况类似.
3 结 论
应用DFT B3LYP和QCISD方法首次研究了三重态类硅烯HB═SiLiF的结构及其与R—H(R= F,OH,NH2)的插入反应.构型优化计算表明三重态的HB═SiLiF有三种平衡构型,其中四元环结构能量最低,是其存在的主要构型.HB═SiLiF与HF, H2O和NH3发生插入反应的机理相同:反应历程均经过一个带有三元环结构部分的过渡态和一个中间体到达产物.相同条件下,HF最容易与HB═SiLiF发生插入反应,H2O次之,NH3则最难.本文结果进一步丰富了类硅烯的研究内容,并为实验上寻找三重态类硅烯提供了理论参考信息.
1 Tamao,K.;Kawachi,A.Angew.Chem.Int.Edit.,1995,34:818
2 Lee,M.E.;Cho,H.M.;Lim,Y.M.;Choi,J.K.;Park,C.H.;Jeong, S.E.;Lee,U.Chem.Eur.J.,2004,10:377
3 Molev,G.;Bravo-Zhivotovakii,D.;Karni,M.;Tumanskii,B.; Botoshansky,M.;Apeloig,Y.J.Am.Chem.Soc.,2006,128:2784
4 Boudjouk,P.;Samaraweera,U.Angew.Chem.Int.Edit.,1988,27: 1355
5 Tamao,K.;Kawachi,A.Organometallics,1995,14:3108
6 Tamao,K.;Kawachi,A.Pure Appl.Chem.,1999,71:393
7 Sekiguchi,A.;Lee,V.Y.;Nanjo,M.Coord.Chem.Rev.,2000, 210:11
8 Wiberg,N.;Niedermayer,W.J.Organomet.Chem.,2001,628:57
9 Likhar,P.R.;Zirngast,M.;Baumgartner,J.;Marschner,C.Chem. Commun.,2004:1764
10 Kawachi,A.;Oishi,Y.;Kataoka,T.;Tamao,K.Organometallics, 2004,23:2949
11 Lim,Y.M.;Cho,H.M.;Lee,M.E.;Baeck,K.K.Organometallics, 2006,25:4960
12 Harloff,J.;Popowski,E.;Reinke,H.J.Organomet.Chem.,2007, 692:1421
13 Clark,T.;Schleyer,P.R.J.Organomet.Chem.,1980,191:347
14 Feng,S.;Feng,D.;Deng,C.Chem.Phys.Lett.,1993,214:97
15 Tanaka,Y.;Kawachi,A.;Hada,M.;Nakatsuji,H.;Tamao,K. Organometallics,1998,17:4573
16 Feng,S.;Feng,D.;Li,J.Chem.Phys.Lett.,2000,316:146
17 Feng,S.;Feng,D.J.Mol.Struct.-Theochem,2001,514:171
18 Feng,S.;Feng,D.;Li,M.;Bu,Y.Chem.Phys.Lett.,2001,339: 103
19 Feng,S.;Feng,D.;Li,M.;Zhou,Y.Int.J.Quantum Chem.,2002,87:360
20 Feng,S.;Zhou,Y.;Feng,D.J.Phys.Chem.A,2003,107:4116
21 Feng,D.;Xie,J.;Feng,S.Chem.Phys.Lett.,2004,396:245
22 Feng,S.;Lai,G.;Zhou,Y.;Feng,D.Chem.Phys.Lett.,2005,415: 327
23 Flock,M.;Marschner,C.Chem.Eur.J.,2005,11:4635
24 Xie,J.;Feng,D.;Feng,S.;Zhang,J.J.Mol.Struct.-Theochem, 2005,755:55
25 Xie,J.;Feng,D.;He,M.;Feng,S.J.Phys.Chem.A,2005,109: 10563
26 Xie,J.;Feng,D.;Feng,S.J.Organomet.Chem.,2006,691:208
27 Xie,J.;Feng,D.;Feng,S.Struct.Chem.,2006,17:63
28 Xie,J.;Feng,D.;Feng,S.J.Comput.Chem.,2006,27:933
29 Xie,J.;Feng,D.;Feng,S.;Zhang,J.Chem.Phys.,2006,323:185
30 Li,W.Z.;Gong,B.A.;Cheng,J.B.Acta Phys.-Chim.Sin.,2006, 22:653 [李文佐,宫宝安,程建波.物理化学学报,2006,22: 653]
31 Li,W.Z.;Gong,B.A.;Cheng,J.B.;Xiao,C.P.Acta Chim.Sin., 2007,65:1573 [李文佐,宫宝安,程建波,肖翠平.化学学报, 2007,65:1573]
32 Xie,J.;Feng,D.;Feng,S.;Ding,Y.Struct.Chem.,2007,18:65
33 Xie,J.;Feng,D.;Feng,S.J.Phys.Chem.A,2007,111:8475
34 Qi,Y.;Feng,D.;Feng,S.J.Mol.Struct.-Theochem,2008,856: 96
35 Li,W.Z.;Cheng,J.B.;Gong,B.A.;Yu,J.K.;Sun,J.Z.Acta Phys.-Chim.Sin.,2008,24:901 [李文佐,程建波,宫宝安,于健康,孙家钟.物理化学学报,2008,24:901]
36 Qi,Y.;Feng,D.;Li,R.;Feng,S.J.Organomet.Chem.,2009,694: 771
37 Li,W.Z.;Cheng,J.B.;Gong,B.A.;Yu,J.K.;Sun,J.Z.Acta Chim.Sin.,2009,67:756 [李文佐,程建波,宫宝安,于健康,孙家钟.化学学报,2009,67:756]
38 Holthausen,M.C.;Koch,W.;Apeloig,Y.J.Am.Chem.Soc., 1999,121:2623
39 Jiang,P.;Gaspar,P.P.J.Am.Chem.Soc.,2001,123:8622
40 Hill,N.J.;West,R.J.Organomet.Chem.,2004,689:4165
41 Cramer,C.J.;Falvey,D.E.Tetrahedron Lett.,1994,35:4943
42 Tachikawa,H.;Yamada,Y.;Lyama,T.Can.J.Chem.,1999,77: 1419
43 Gaspar,P.P.;Xiao,M.;Pae,D.H.;Berge,D.J.;Haile,T.;Chen, T.;Lei,D.;Winchester,W.R.;Jiang,P.J.Organomet.Chem., 2002,646:68
44 Yoshida,M.;Tamaoki,N.Organometallics,2002,21:2587
45 Sekiguchi,A.;Tanaka,T.;Ichinohe,M.;Akiyama,K.; Tero-Kubota S.J.Am.Chem.Soc.,2003,123:4962
46 Xu,Y.;Zhang,Y.;Li,J.J.Phys.Chem.C,2007,111:3729
47 Skryshevski,Y.;Piryatinski,Y.;Vakhnin,A.;Blonsky,I.; Kadashchuk,A.;Ne"pu˚rek,S.Optical Materials,2007,30:384
48 Becke,A.D.J.Chem.Phys.,1993,98:5648
49 Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:785
50 Hehre,W.J.;Radom,L.;Schleyer,P.v.R.;Pople,J.A.Ab initio molecular orbital theory.New York:Wiley,1986
51 Gonzalez,C.;Schlegel,H.B.J.Phys.Chem.,1991,95:5853
52 Gauss,J.;Cremer,C.Chem.Phys.Lett.,1988,150:280
53 Salter,E.A.;Trucks,G.W.;Bartlett,R.J.J.Chem.Phys.,1989, 90:1752
54 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision B.03.Pittsburgh,PA:Gaussian Inc.,2003
55 Apeloig,Y.;Pauncz,R.;Karni,M.;West,R.;Steiner,W.; Chapman,D.Organometallics,2003,22:3250
Structures of the Triplet Silylenoid HB═SiLiF and Its Insertion Reactions with R—H(R=F,OH,NH2)
LI Wen-Zuo*ZHU Hong-Jie CHENG Jian-Bo LI Qing-Zhong GONG Bao-An
(Chemistry and Biology College,Yantai University,Yantai 264005,Shandong Province,P.R.China)
Density functional theory(DFT)and quadratic configuration interaction with single and double excitations (QCISD)methods were used to investigate the geometries of the triplet silylenoid HB═SiLiF as well as its insertion reactions with RH(R=F,OH,NH2).The calculated results indicated that HB═SiLiF has three equilibrium structures wherein the four-membered ring structure had the lowest energy and it was the most stable structure.The mechanisms of the insertion reactions for HB═SiLiF with HF,H2O,and NH3were identical.The QCISD/6-311++G(d,p)//B3LYP/ 6-311+G(d,p)calculated potential energy barriers of the three reactions were 124.85,140.67,and 148.16 kJ·mol-1,and the reaction heats for the three reactions were-2.22,20.08,and 23.22 kJ·mol-1,respectively.Under the same conditions,the insertion reactions should occur easily according to the following order:H—F>H—OH>H—NH2.
Insertion reaction;Silylenoid HB═SiLiF;B3LYP;QCISD
O641.12
Received:March 17,2010;Revised:April 6,2010;Published on Web:June 29,2010.
*Corresponding author.Email:liwenzuo2004@126.com;Tel:+86-535-6902063.
The project was supported by the Natural Science Foundation of Shandong Province,China(ZR2009BQ006),Open Fund of the State Key Laboratory
of Supramolecular Structure and Materials(Jilin University,China)(SKLSSM200909),and Fund for Doctor of Yantai University,China(HY05B30, HY05B36).
山东省自然科学基金(ZR2009BQ006),超分子结构与材料国家重点实验室(吉林大学)开放基金(SKLSSM200909)和烟台大学博士科研基金
(HY05B30,HY05B36)资助项目
ⒸEditorial office of Acta Physico-Chimica Sinica
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
陶瓷学报(2020年2期)2020-10-27
电脑知识与技术(2018年3期)2018-03-21
中国资源综合利用(2017年4期)2018-01-22
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
当代化工研究(2016年7期)2016-03-20
股市动态分析(2015年12期)2015-09-10