
HPLC法测定甲氧苄啶片主药及有关物质的含量
2010-09-11 08:05:56池秀珍福建泉州市药品检验所泉州市362000
中国药房 2010年5期
池秀珍(福建泉州市药品检验所,泉州市 362000)
甲氧苄啶为一种广谱、高效、低毒的抗菌增效剂[1],其制剂甲氧苄啶片收载于《中国药典》2005年版二部中。该标准[2]中含量测定采用紫外分光光度法,其原料药项下有关物质检查采用薄层色谱法,但甲氧苄啶片项下未对有关物质进行控制。为了更全面的控制甲氧苄啶片的质量,笔者建立了高效液相色谱(HPLC)法同时测定甲氧苄啶片中主药及有关物质的含量。结果表明,该法简便、准确,专属性强,可用于甲氧苄啶片的含量测定及有关物质的检查。
1 仪器与试药
1100系列HPLC仪、色谱工作站(美国Agilent公司)。
甲氧苄啶标准品(中国药品生物制品检定所,批号:100031-200304,供含量测定用);甲氧苄啶片(广东台城制药有限公司,批号:20081001、20090402;江苏永大药业有限公司,批号:20060303。规格均为每片100 mg);乙腈为色谱纯,磷酸、三乙胺均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件及系统适用性试验
色谱柱:Waters symmetry C18(250 mm×4.6 mm,5 μm);流动相:0.015 mol·L-1磷酸(用三乙胺调pH至3.5)-乙腈(83∶17);流速:1 mL·min-1;柱温:30 ℃;检测波长:271 nm;进样量:10 μL。取“2.2”项下标准品溶液、供试品溶液与阴性对照溶液进样分析,结果在上述色谱条件下,理论板数按甲氧苄啶计为11 053,阴性对照溶液不干扰测定。色谱见图1。
2.2 溶液的制备

图1 高效液相色谱图Fig 1 HPLC chromatography
2.2.1 标准品溶液。精密称取甲氧苄啶标准品(105℃干燥3 h)20 mg置于25 mL容量瓶中,加0.01 mol·L-1HCl溶液适量,超声使溶解,稀释至刻度,摇匀,作为标准品贮备液。精密吸取贮备液5 mL,置于50 mL容量瓶中,用0.01 mol·L-1HCl溶液稀释至刻度,摇匀。
2.2.2 供试品溶液与阴性对照溶液。取本品20片,精密称定,研细,精密称取适量(约相当于甲氧苄啶80 mg)置于100 mL容量瓶中,加0.01 mol·L-1HCl溶液适量,超声使溶解,并稀释至刻度,摇匀,滤过。精密吸取续滤液5 mL,置于50 mL容量瓶中,加0.01 mol·L-1HCl溶液至刻度,摇匀,作为供试品溶液;另按处方比例称取空白辅料适量,同法制成阴性对照溶液。
2.3 专属性试验
分别取本品细粉适量,分置于4个10 mL具塞试管中,1份加入0.01 mol·L-1HCl溶液10 mL摇匀后于4 500 Lx光照48 h;1份加3%H2O2溶液10 mL摇匀后于80℃水浴1 h;另2份分别加入1 mol·L-1HCl溶液10 mL、3 mol·L-1NaOH溶液10 mL于80℃水浴2 h。精密吸取各条件下破坏后的样品溶液各1 mL,分置于10 mL容量瓶中(经酸、碱破坏的样品在稀释前先中和),加0.01 mol·L-1HCl溶液至刻度,摇匀。取上述各溶液,照“2.1”项下的色谱条件,分别进样10 μL,记录色谱至主峰保留时间的4倍。结果,供试液在氧化破坏条件下产生的降解产物峰最多,其他破坏条件下也均有不同程度的降解产物产生。在此色谱条件下,甲氧苄啶与其相邻降解产物峰都能达到有效分离,降解产物不干扰样品的测定,色谱见图1。
2.4 线性关系考察
精密吸取标准品贮备液0.2、0.6、1.0、1.4、2.0 mL分别置于10 mL容量瓶中,用0.01 mol·L-1HCl溶液稀释至刻度,摇匀,得系列标准品溶液。分别取上述溶液各10 μL注入色谱仪,照“2.1”项下色谱条件测定,记录色谱。以标准品的浓度(C)为横坐标,峰面积(Y)为纵坐标进行线性回归,得线性方程为Y=11.658 6C+4.298 7(r=0.999 9,n=5)。结果表明,甲氧苄啶检测浓度线性范围为16.1~161.3 μg·mL-1。
2.5 精密度试验
取“2.4”项下标准品溶液(80 μg·mL-1),连续进样5次,测定峰面积,结果峰面积的RSD=0.1%。
2.6 重复性试验
取同一批样品(20081001)6份,照“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件试验,测得其平均含量为99.6%,RSD=0.3%。
2.7 稳定性试验
取“2.6”项下的供试品溶液,室温放置,分别于0、2、4、8、12、24 h时进样10 μL,结果其峰面积的RSD=0.5%。表明供试品溶液在室温放置24 h内稳定。
2.8 加样回收率试验
取已知含量的样品9份,按高、中、低浓度加入不同量的标准品各3份,照“2.1”项下色谱条件测定,计算回收率,结果见表1。
2.9 最低检测限与定量限
取标准品溶液逐级稀释、测定,按信噪比(S/N)为3∶1,测得最低检测限为3.8 ng;按信噪比(S/N)为10∶1,测得定量限为13.9 ng。
2.10 样品中主药及有关物质含量测定
取不同生产厂家的样品3批,按“2.2”项下方法制备供试品溶液、标准品溶液,照“2.1”项下色谱条件测定,记录色谱,按外标法以峰面积计算含量,并与《中国药典》记载的紫外分光光度法测定结果进行比较。
精密称取本品细粉适量,用0.01 mol·L-1HCl溶液制成每1 mL含0.8 mg的溶液,滤过,取续滤液作为供试液;精密吸取供试液1 mL,用0.01 mol·L-1HCl溶液制成每1 mL含0.008 mg的溶液作为对照溶液。取对照溶液10 μL,注入色谱仪,调节仪器检测灵敏度,使主成分色谱峰的峰高约为满量程的20%~25%,再精密吸取供试液与对照溶液各10 μL,分别注入色谱仪,记录色谱至主成分峰保留时间的2倍,供试液如有杂质峰,按主成分自身对照法计算杂质总量,各杂质峰面积的和不得大于对照溶液主峰面积(1.0%)。
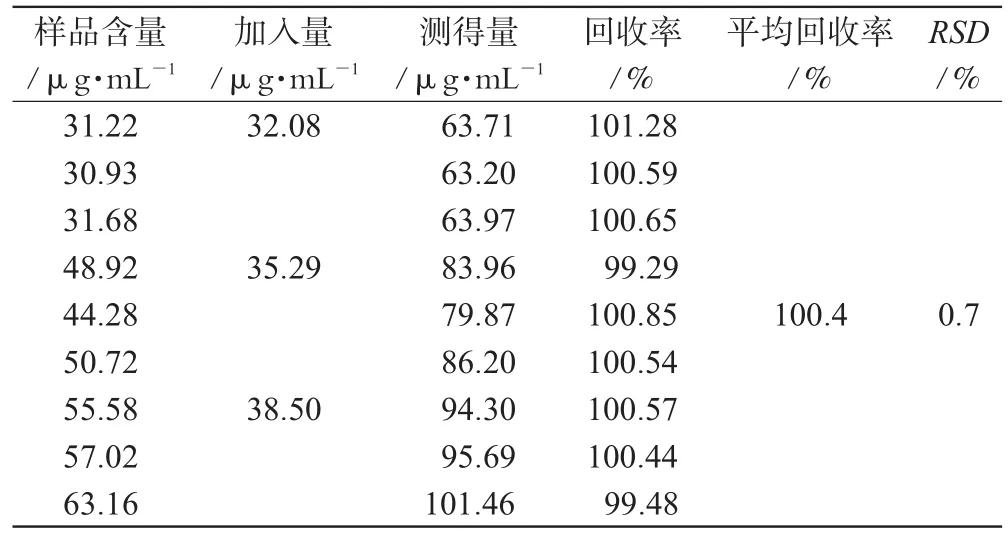
表1 回收率试验结果(n=9)Tab 1 Result of recovery test(n=9)
主药及有关物质含量测定结果见表2。
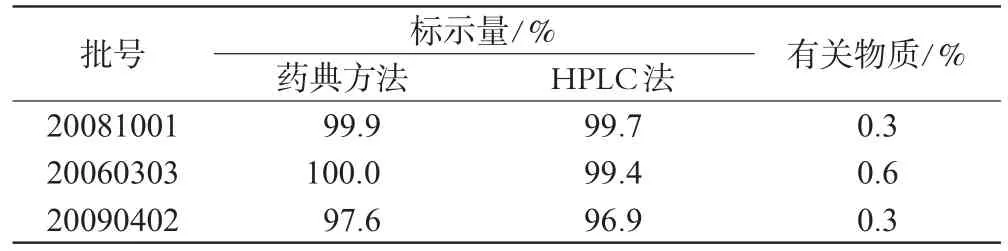
表2 样品中主药及有关物质含量测定结果(%%)Tab 2 Content determination of trimethoprim and its related substances(%%)
由表2可见,本文建立的主药含量测定方法与药典法比较结果无差异。
3 讨论
笔者参考了相关文献[3~5],比较了不同比例、不同pH的乙腈-磷酸溶液以及甲醇-0.1 mol·L-1磷酸二氢钾溶液作为流动相系统时的分离效果。结果以0.015 mol·L-1磷酸(用三乙胺调pH至3.5)-乙腈(83∶17)为流动相时,甲氧苄啶色谱峰峰形最好,保留时间适中(5.7 min),主峰与其相邻的杂质峰能有效分离,溶剂、辅料均不干扰测定。
[1]张 伦.甲氧苄啶的应用、生产和市场状况[J].中国药房,2001,12(3):143.
[2]国家药典委员会编.中华人民共和国药典(二部)[S].2005年版.北京:化学工业出版社,2005:118.
[3]国家药典委员会编.国家药品标准化学药品[S].第十三册.2002,12:108.
[4]韩志云,朱迎春.HPLC法测定头孢氨苄甲氧苄啶胶囊含量的方法改进[J].海峡药学,2007,19(1):48.
[5]谢 燕,陈 明,肖艳萍.高效液相色谱法测定甲氧苄啶氯化钠注射液中甲氧苄啶的含量[J].精细化工中间体,2006,36(4):63.
猜你喜欢
中学生数理化·自主招生(2022年4期)2022-05-09 22:00:23
家庭医学(下半月)(2020年7期)2020-04-18 13:45:31
中学化学(2019年4期)2019-08-06 13:59:37
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06 07:32:18
数学小灵通(1-2年级)(2017年9期)2017-10-13 08:10:20
学苑创造·B版(2017年1期)2017-02-21 18:52:47
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
北方牧业(2016年1期)2016-12-17 19:08:50
小天使·二年级语数英综合(2016年9期)2016-05-14 13:03:38
中国医药生物技术(2014年4期)2014-01-23 09:24:24
