
M’型GdTaO4电子结构的第一性原理研究*
2010-09-08 06:05顾牡林玲刘波刘小林黄世明倪晨
物理学报 2010年4期
顾牡林玲 刘波 刘小林 黄世明 倪晨
(同济大学物理系,上海市特殊人工微结构材料与技术重点实验室,上海200092)
(2009年6月5日收到;2009年8月17日收到修改稿)
M’型GdTaO4电子结构的第一性原理研究*
顾牡†林玲 刘波 刘小林 黄世明 倪晨
(同济大学物理系,上海市特殊人工微结构材料与技术重点实验室,上海200092)
(2009年6月5日收到;2009年8月17日收到修改稿)
运用基于密度泛函理论的赝势平面波方法计算了M’型GdTaO4的电子结构.结果表明:M’型GdTaO4价带顶主要由O-2p电子构成,导带底由Ta-5d的e轨道电子构成;当Ueff=8 eV时,自旋向上和自旋向下的Gd-4f电子分别局域于价带顶以下6.27 eV和导带底以上3.01 eV处;计算得到M’型GdTaO4的折射率为2.24,与应用半经验的Gladstone-Dale关系得到的结果符合得很好.
M’型钽酸钆,第一性原理计算,能带,态密度
PACC:7850E,3120A,7125C,7120H
1. 引言
钽酸钆(GdTaO4)晶体具有密度大(8.84 g/cm3)、X射线吸收能力强、辐照硬度高、化学性质稳定等特点,该晶体经稀土掺杂可显现出良好的发光性能[1—3],因而是一种十分有效的X射线发光基质材料.采用溶胶凝胶法,我们已经成功制备出了高质量的GdTaO4:Eu3+薄膜,并对其发光性质进行了研究[4,5].为了分析有关的实验现象,进一步改善材料的发光性能,需要了解GdTaO4晶体的电子结构,然而目前有关GdTaO4晶体电子结构的报道十分缺乏.为此本文采用第一性原理计算对GdTaO4晶体的电子结构进行了研究.
镧系钽酸盐(MTaO4)晶体可以由M2O3和Ta2O5在特定的温度下混合烧制而成.由于烧结温度和镧系离子M3+半径大小的不同,将形成不同的晶相结构. GdTaO4晶体主要可形成两种单斜结构:第一种群号为P2/a(M’型),每个晶胞中含有两个GdTaO4分子,如图1(a),其中O有两种格位,分别表示为O1和O2[6],Ta离子位于6个O离子所构成的畸变八面体中,形成TaO6基团,如图1(b)所示,这些基团相互连接,这样Gd原子就位于8个O原子形成的不同的Ta—O环境中如图1(c)所示[7];第二种群号为I2/a (M型),每个晶胞中含有四个GdTaO4分子[8,9].我们制备出的GdTaO4:Eu3+薄膜为M’型晶相[4],该结构在室温下稳定,且发光性能优于M型[10],因此本文的研究主要针对M’型GdTaO4晶体展开.
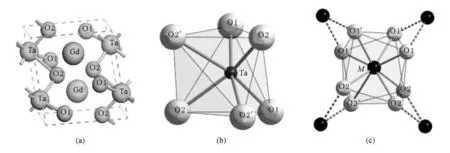
图1 M’型GdTaO4的晶体结构(a)M’型GdTaO4晶体结构,(b)TaO6基团,(c)MO8基团
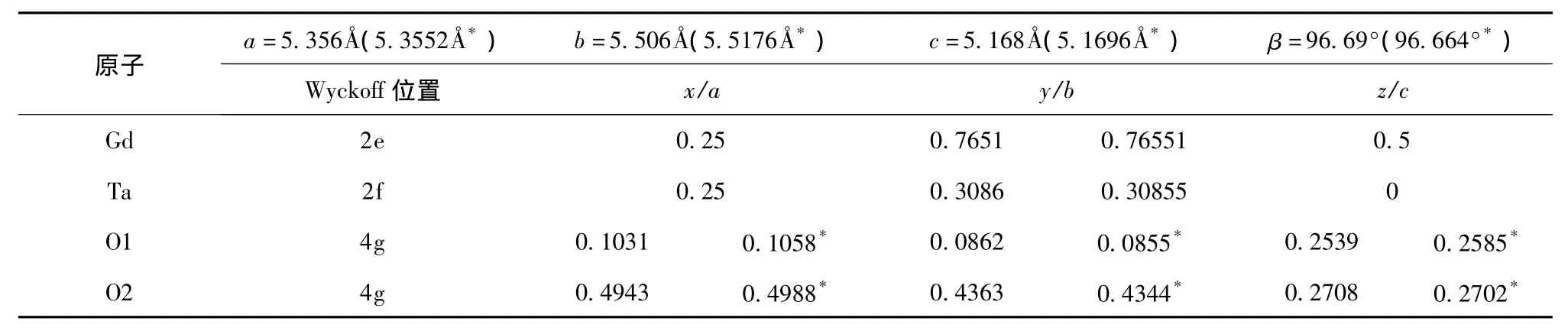
表1 M’型GdTaO4的晶格常数及原子位置[7,11]
2. 计算方法
基于赝势平面波方法,采用VASP(Vienna ab initio simulation package)程序包[12]对M’型GdTaO4的电子结构等性质进行了第一性原理计算.在构造投影扩展平面波(PAW)势[13]时,选取Gd的5s25p64f75d16s2,Ta的5d36s2和O的2s22p4电子为价电子;交换关联势选取Perdew,Burke和Ernzerehof提出的基于广义梯度近似(GGA)的PBE势[14];平面波基集的截断能设为520 eV;自洽迭代过程采用Monkhorst-pack的方法生成以Γ点为中心的6×6×6 k点网格,通过几何优化使得每个原子上的剩余力小于0.01 eV/.
由于Gd原子的4f壳层含有半满的7个电子,因此应当将GdTaO4作为强关联材料进行处理,目前较普遍且有效的是采用考虑在位Coulomb能的密度泛函理论(DFT+U)[15,16]的方法:将电子分成两个子系统,用L(S)DA/GGA处理s和p电子,对d和f电子在Hamilton中考虑在位Coulomb能U的作用.U值代表的物理意义是把一个d或f电子放到另一同种原子相同轨道上体系前后的能量差.
为了正确地计算GdTaO4的基态性质,必须在平均场近似下考虑Gd-4f电子的在位Coulomb能U.U作为一个半经验参数,需要磁矩、带隙、体模量,Gd-4f电子占据态与非占据态的能量差等实验值的比对验证[17,18],但是GdTaO4相关的实验数据十分匮乏,增加了确定U值的难度.然而,另一方面镧系离子的4f电子受到5s25p6壳层的屏蔽[19],因此周围晶体场对其的作用相当有限,它们的诸多性质(如:磁矩,光谱等)往往表现出单原子或离子的特性[19].由此,通过比照其他含Gd化合物中4f电子的性质可为推测GdTaO4中的U值提供参考.
本文选择Dudarev提出的有效在位Coulomb能Ueff=U-J方法[20]进行计算,采用交换参数J=1 eV,通过研究不同Ueff值(0—8 eV)对GdTaO4带隙,Gd-4f电子非占据态与占据态能量差的影响,推测Ueff的大小,并以此计算了M’型GdTaO4的电子结构.
3. 结果和讨论
3.1. 有效在位Coulomb能Ueff
就Gd-4f电子非占据态与占据态的能量差而言,实验发现:Gd金属单质为12 eV[16],GdN为13 eV[21],GdBi为13.4 eV,GdSb为13.3 eV,GdAs为13.1 eV,GdP为13.2 eV[22].Pidol等采用Dorenbos模型算得在Lu2Si2O5中Gd3+的4f电子非占据态与占据态的能量差为12.8 eV[23].根据已有的结果,材料中Gd-4f电子非占据态与占据态的能量差通常在12—13.5 eV之间.由于Gd3+的4f电子在5s25p6电子的屏蔽下,基本不参与成键,不同材料中的差别不是很大.
当Ueff=0 eV时,Gd的非占据4f轨道处在TaO4基团的光学禁带中;随着Ueff值的增加,自旋向下的电子能量上移,自旋向上的电子能量下移,Gd-4f电子非占据态与占据态的劈裂增大,能量差的变化基本满足线性关系(如图2),这与Gd金属单质中的情况一致[17].当Ueff=6 eV时,其能量差为11.39 eV,Ueff=8 eV时,能量差为13.31 eV.
从带隙方面来看,钽酸盐的光学吸收带源自O-2p到Ta-5d的电荷转移跃迁[2,24,25],其吸收峰位于220nm(5.64 eV)[26].若Ueff过小,Gd-4f能带将位于禁带之内,与实验不符[24].图3为计算所得Ueff对禁带宽度的影响,随着Ueff值的增大,禁带宽度将逐渐增大,当Ueff≥6 eV时,禁带宽度不再发生变化,约为4.03 eV.这时价带顶主要由O-2p电子构成,导带底主要由Ta-5d构成,与实验结果相符[24,25].计算得到的带隙宽度与实验结果相比存在一定的偏差,这主要源自于密度泛函理论算得的强关联体系禁带宽度会普遍偏小[27,28]所致.
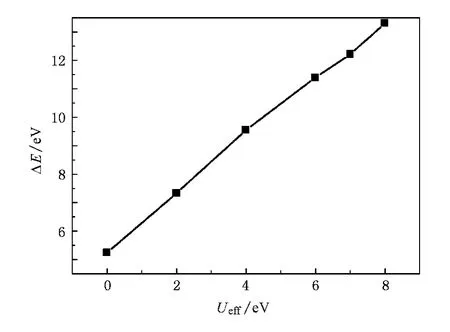
图2 Ueff对Gd-4f电子非占据态与占据态能量差的影响
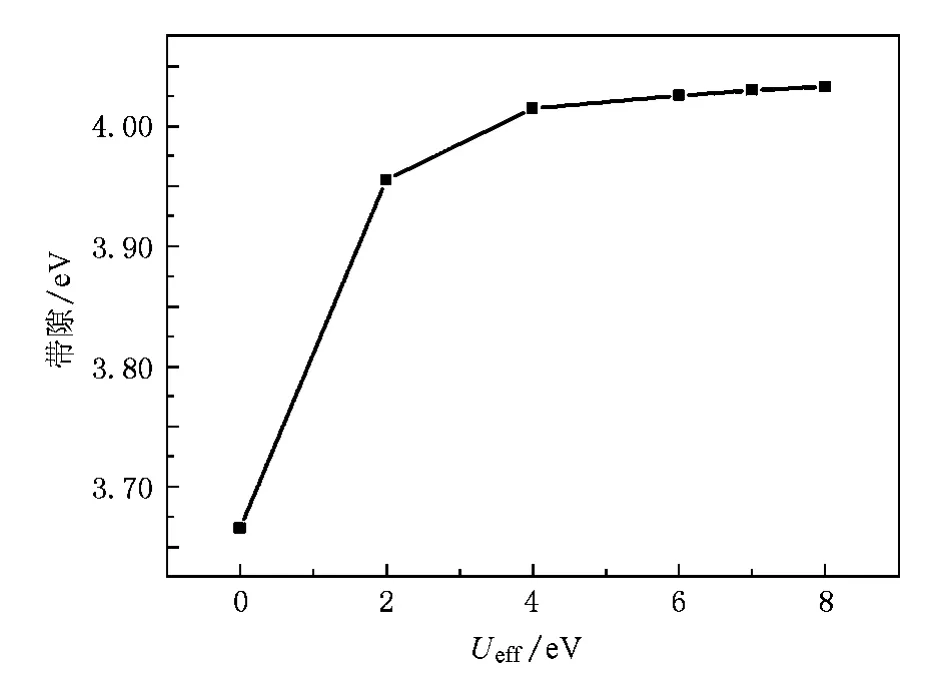
图3 Ueff对禁带宽度的影响
综上所述,我们推测Ueff取值在6.5—8.5 eV之间.鉴于Ueff=8 eV时,几何优化的结果与实验值最为接近,以下取Ueff=8 eV为例,对M’型GdTaO4晶体的电子结构进行计算和分析.
3.2. 态密度
图4为M’型GdTaO4的总态密度和各原子的分态密度图.把价带顶设为0 eV,结合图4(b)的分波态密度图可以看出,图4(a)中的态密度大致可分成三个部分:从-22到-15 eV能区为芯带,主要由Gd-5p和O-2s电子构成;从-7到0 eV能区为价带,主要由O-2p电子构成,在价带低能端还有Gd的局域的4f电子;4—14 eV能区为导带,主要由Ta-5d和Gd-4f电子构成.
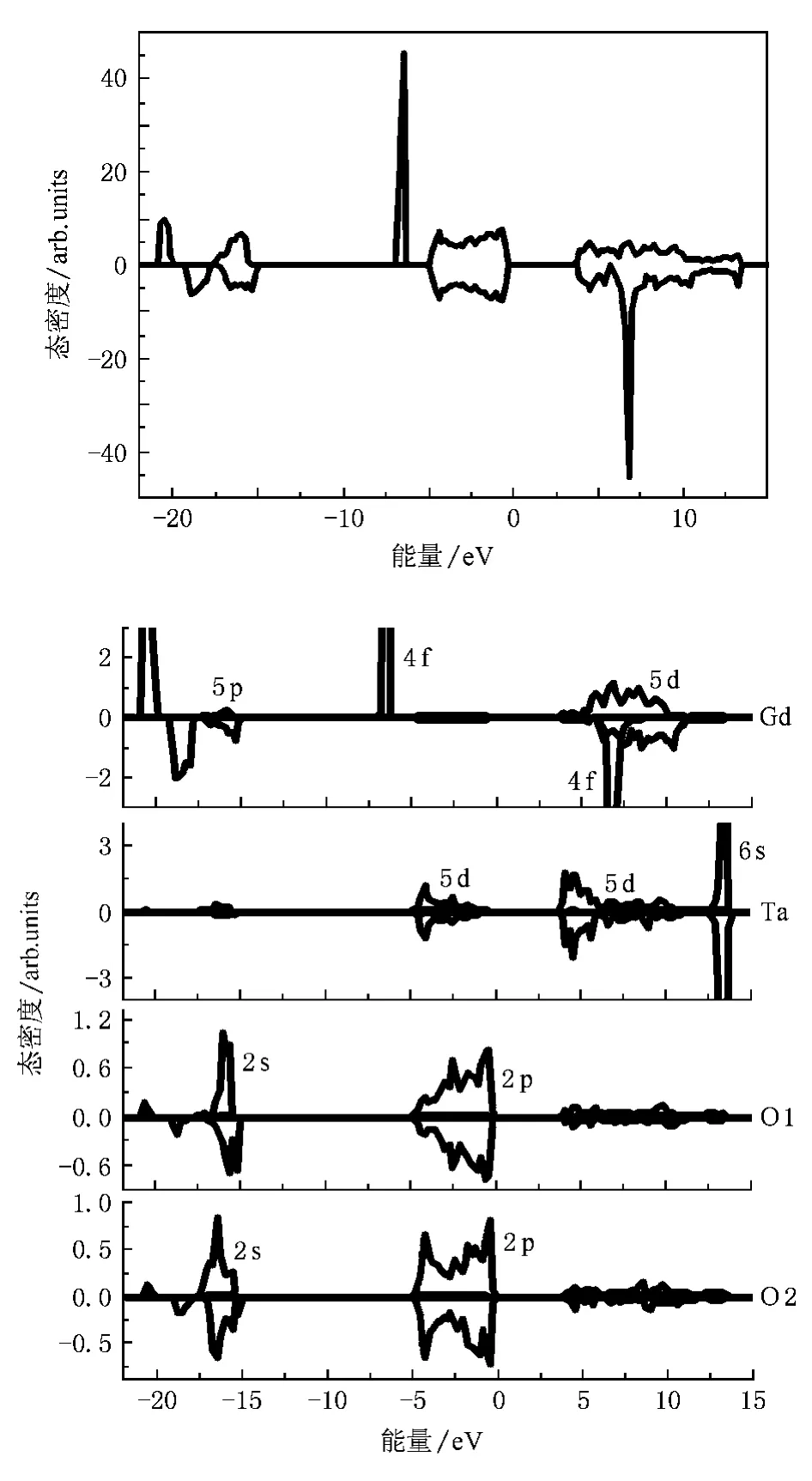
图4 M’型GdTaO4晶体的态密度(a)M’型GdTaO4总态密度,(b)M’型GdTaO4分波态密度
由分态密度图4(b)还可看出两种格位的O离子态密度分布差不多,这是因为O1和O2与其他离子有相似的间距,这个结果与InTaO4的O1和O2情况相同[29].前文提到GdTaO4的光学吸收是O-2p至Ta-5d的电荷转移,O-2p的态密度比较扩展,一个电子从基态的成键轨道跃迁到激发态的反键轨道,发生了很大的电子重组[30],因此实验上测得的TaO4基团的电荷转移激发峰很宽.在GdTaO4:Eu3+材料中,Ta-O之间的电荷转移跃迁能量(220nm)可以直接传递给Eu3+发光,也可以通过Gd3+再传递给Eu3+[2].
为了进一步研究Ta-O之间的相互作用,分别画出了Ta-5d电子的t2和e轨道以及O-2p电子的σ和π轨道的态密度图(见图5).设价带顶为0 eV,从图5中可以发现价带顶O-2p电子π轨道的贡献大于σ轨道的,导带底主要是Ta-5d的e轨道电子的贡献,导带顶主要由t2轨道电子构成.
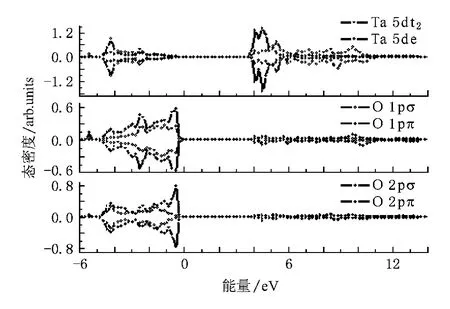
图5 Ta-5d电子的e和t2轨道以及O-2p电子的σ和π轨道态密度
3.3. 能带结构
取群号为P2/a单斜结构的第一Brillouin区中高对称点计算出各个点的能量值,做出M’型GdTaO4晶体的能带结构(如图6).设价带顶为能量零点,图6(a)为自旋向上的能带,图6(b)为自旋向下的能带.图6中可以看出GdTaO4价带顶处在Γ点,导带底处在E-C方向,能带具有间接带隙结构.自旋向上的能带结构中,带隙的计算值为3.99 eV,自旋向下的能带结构中,带隙的计算值为4.07 eV,其平均值为4.03 eV.
从图6中可以看出导带顶端有类抛物线状的能带,结合分波态密度图4(b)可以得出这是来自Ta-6s电子的贡献;两图中均有若干条能量非常局域的能带,图6(a)中距价带顶以下6.27 eV处为自旋向上的Gd-4f电子,图6(b)中距导带底以上3.01 eV处为自旋向下的Gd-4f电子,Gd-4f自旋向上和向下的电子能量差为13.31 eV.
3.4. 电荷密度
取Ta和O1,O2原子所在面做电荷密度图.虽然Gd是在平行于这个面以上0.47376的位置上,但是从价带底和导带顶的图中也能看到Gd的电荷密度,它的坐标为(-0.17945,3.50750);设Ta原子的位置为坐标零点,则O2坐标为(-1.67238,1.44316),O1为(1.03924,1.57408);在0.02—0.1 e范围内取电荷密度间距为0.02 e/3,在范围为0.1—6 e内取间距为0.1 e/3.
Gd-4f电子在5s5p电子的屏蔽下,受晶体场影响较小,与其他电子的杂化较小.图7中价带底能量范围为从-7到-3 eV,有密集的Gd-4f电荷密度,与O原子几乎没有杂化作用;Ta-5d的t2轨道电子占主导地位,在与O原子的作用中,看不出明显的σ键和π键特征.在从-3到0 eV的价带顶能量范围内,Ta-5d电子与O-2p电子的作用中主要以π键为主导.导带底能量范围为从4到5 eV,Ta-5d电子的e轨道电荷密度十分明显,O的电荷密度明显减小.在从5到14 eV能量范围的导带顶,基本看不到Ta-5d电子的电荷密度,Gd-4f的电荷密度十分明显.这些结果与态密度的结果一一对应.
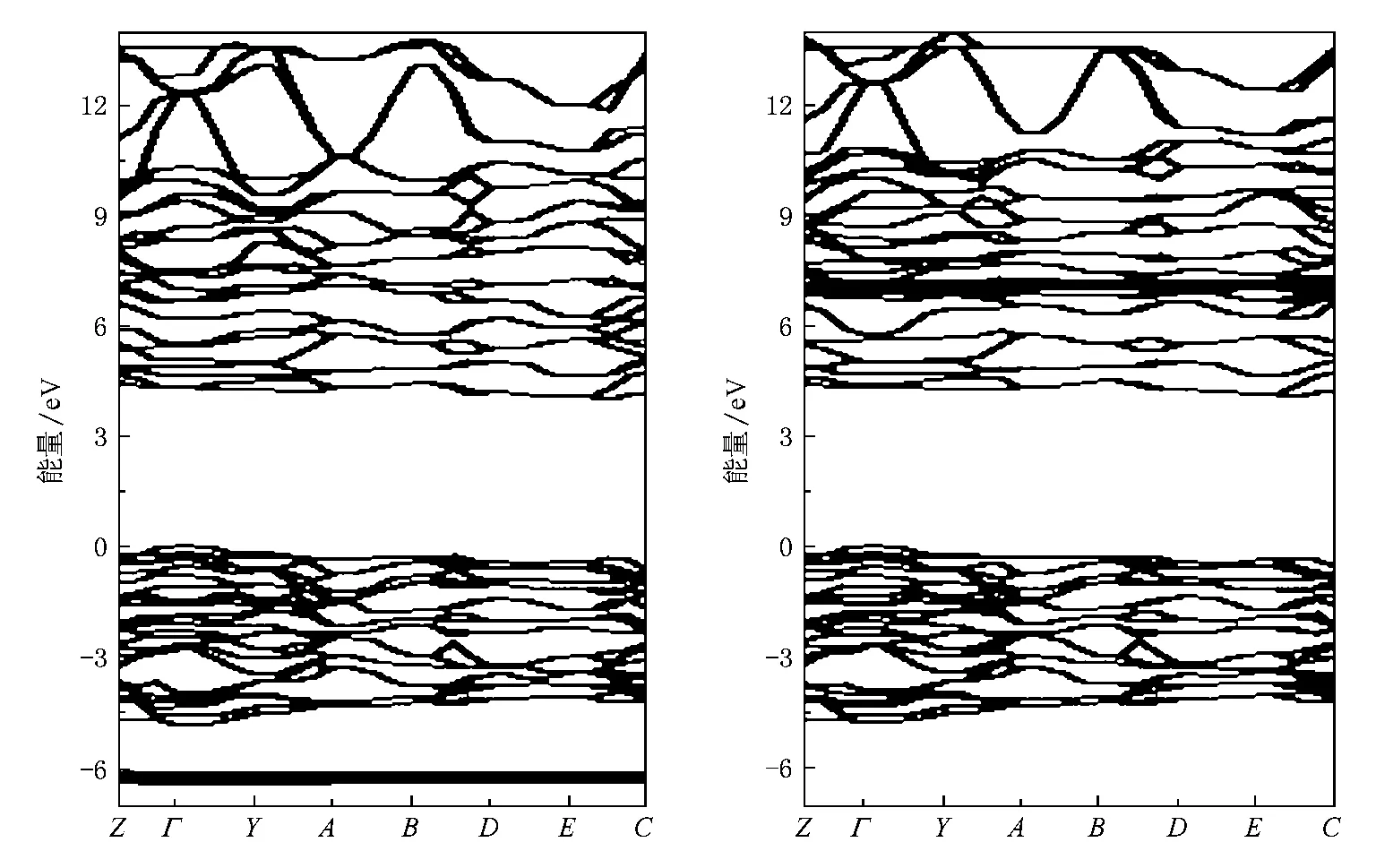
图6 M’型GdTaO4能带结构(a)为自旋向上能带,(b)为自旋向下能带
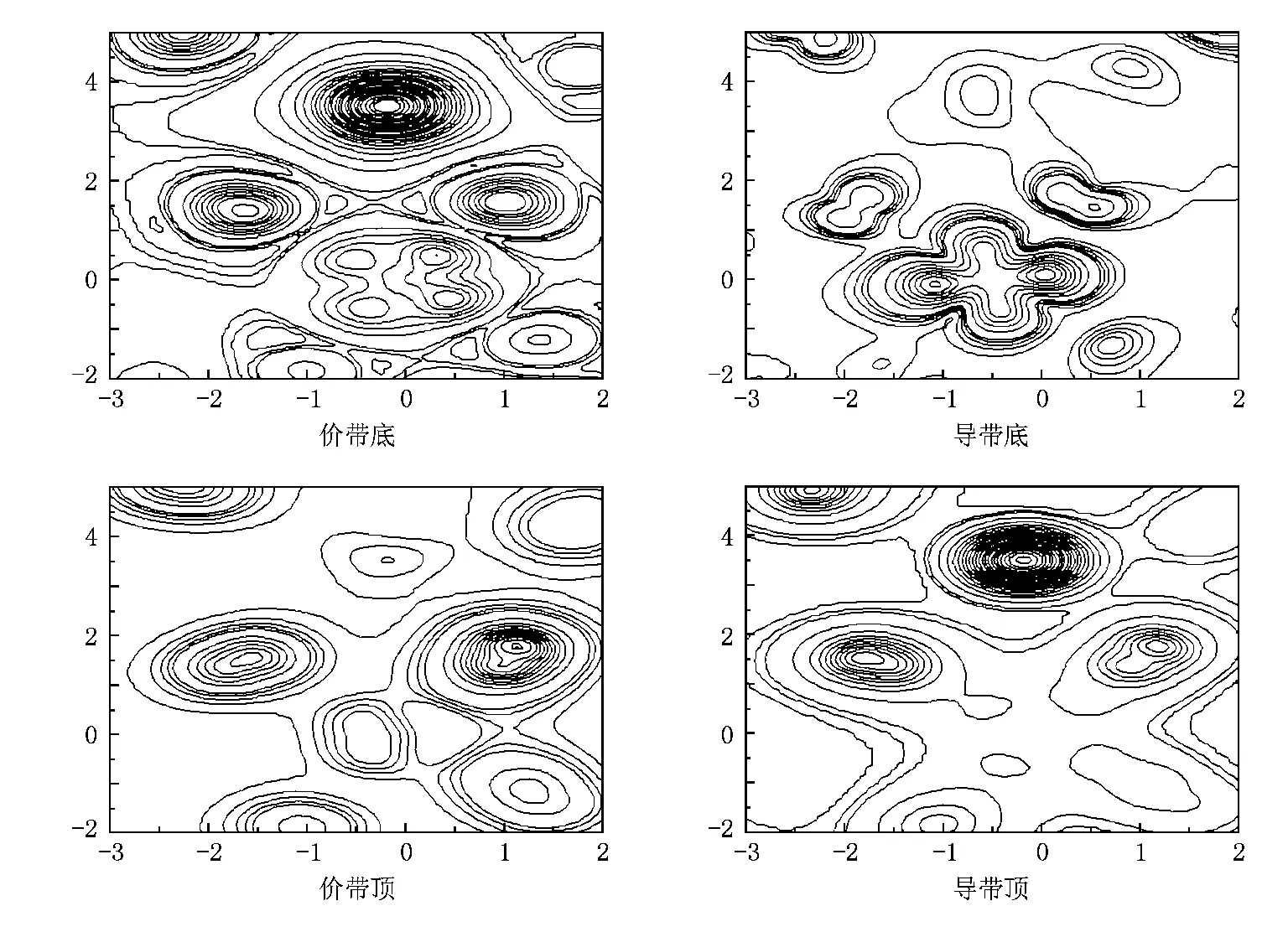
图7 Ta-O1-O2面电荷密度
3.5. 折射率
利用Sternheimer理论[31],我们还计算出M’型GdTaO4的静态介电常数ε.并由折射率n与介电常数ε的关系[32,33]得到材料的折射率(表2).虽然M’型GdTaO4的折射率是各向异性的,由于x,y,z三个方向差别不大,取它们的平均值2.24作为折射率的计算值,该值与应用半经验Gladstone-Dale关系得到的折射率(n=2.22)符合得很好[34],说明我们的计算是可靠的.

表2 M’型GdTaO4静态介电常数ε和折射率n
4. 结论
通过上述计算和分析可以得到以下结论:1) M’型GdTaO4价带顶主要由O-2p电子构成,导带底由Ta-5d的e轨道电子构成;晶体中Gd-4f电子非占据态与占据态的能量差随Ueff增加线性递增,当Ueff≥6 eV,禁带宽度将不再发生变化,约为4.03 eV; 2)由于受5s和5p电子的屏蔽,晶体场对Gd-4f电子的影响很小,当Ueff=8 eV时,自旋向上和自旋向下的Gd-4f电子将分别局域于价带顶以下6.27 eV和导带底以上3.01 eV处;3)M’型GdTaO4的折射率为2.24,此结果与应用Gladstone-Dale关系得到的结果符合.
这一结果较为清晰地揭示了M’型GdTaO4晶体的电子结构,对于深入探讨稀土掺杂M’型GdTaO4晶体的能量传递过程和发光机制,不断改善材料的发光性能具有积极意义.
[1]Liu G K,Bernard J 2005 Spectroscopic Properties of Rare Earths in Optical Materials(Beijing:Tsinghua University Press)p504
[2]Liu B,Han K,Liu X L,Gu M,Huang S M,Ni C,Qi A M,Zhang G B 2007 Solid State Commun.144 484
[3]Li B,Gu Z N 2000 J.Mater.Sci.35 1139
[4]Gu M,Xiao L H,Liu X L,Zhang R,Liu B J,Xu X 2006 J. Alloys Compd.426 390
[5]Liu X L,Xu X,Gu M,Xiao L H,Han K,Zhang R 2007 Appl. Surf.Sci.253 4344
[6]Keller C 1962 Z.Anorg.Allg.Chem.318 89
[7]Ingo H,Falk L,Tanja N,Steffen F M,Helge M B,Thomas S
2005 Z.Anorg.Allg.Chem.631 2377
[8]Wolten G M 1967 Acta Crystallogr.23 938
[9]Kern W 1978 Radio Corp.Am.Rev.39 278
[10]Brixner L H,Chen H Y 1983 J.Electrochem.Soc.130 2435
[11]Trunov V K,Kinzhibalo L N,Efremov V A,Krongauz V G 1981 Donklady Akademii Nauk SSR 260 103
[12]Kresse G,Furthmuller J 1996 Phys.Rev.B 54 11169
[13]Kresse G,Joubert J 1999 Phys.Rev.B 59 1758
[14]Perdew J P,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[15]Anisimov V I,Aryasetiawan F,Lichtenstein A I 1997 J.Phys. Condens.Matt.9 767
[16]Anisimov V I,Zaanen J,Andersen O K 1991 Phys.Rev.B 44 943
[17]Melissa P,Jurgen H,Martijn M 2006 J.Phys.Condens.Matt. 18 7021
[18]Zhang J H,Liu G,Gu F,Liu L J,Liu M 2006 Acta Phys.Sin. 55 2928(in Chinese)[张加宏、刘甦、顾芳、杨丽娟、刘楣2006物理学报55 2928]
[19]Efthimios K 2007 Atomic and Electronic Structure of Solids (Beijing:World Book Publishing Company)p241
[20]Perdew J P,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[21]Antonov V N,Harmon B N,Yaresko A N,Shpak A P 2007 Phys.Rev.B 75 184422
[22]Hajime Y,Tadashi F,Takayuki M,Yoshiki T,Shin I,Shigemass S,Li D X,Takashi S 1996 J.Phys.Soc.Jpn.65 1000
[23]Pidol L,Viana B,Galtayries A,Dorenbos P 2005 Phys.Rev.B 72 125110
[24]Blasse G,Bril A 1970 J.Lumin.3 109
[25]Blasse G,Brixner L H 1990 Chem.Phys.Lett.173 409
[26]Blasse G,Dirksen G J 1994 J.Alloys Compd.209 1
[27]Hyuniu C,Kijeong K,Yong S C,Eunjeong I,Youngmin C,Jin O B,Sang J M 2004 Chem.Phys.Lett.398 449
[28]Xu X F,Shao X H 2009 Acta Phys.Sin.58 1908(in Chinese)[徐新发、邵晓红2009物理学报58 1908
[29]Shigenori M,Kenji O,Hiroyuki N,Masao A,Kenkichiro K 2003 J.Phys.Chem.Solids 64 2417
[30]BlasseC,GrabmaierBC1994LuminescentMaterials (Germany:Springer-Verlag Berdelberg)p53
[31]Gajos M,Hummer K,Kresse G,Furmuller J,Bechstedt F 2006 Phys.Rev.B 73 045112
[32]Zhang J H,Ding J W,Lu Z H 2009 Acta Phys.Sin.58 1901 (in Chinese)[张计划、丁建文、卢章辉2009物理学报58 1901]
[33]Karazhanov S Z,Ravindran P,Kjekshus A,Fjellvag H,Svensson B G 2007 Phys.Rev.B 75 155104
[34]Mandarino J A 1976 Can.Mineral.14 498
PACC:7850E,3120A,7125C,7120H
*Project supported by the National Natural Science Foundation of China(Grant Nos.50672068,10875085).
†E-mail:mgu@tongji.edu.cn
Fist-principle calculation for electronic structure of M’-GdTaO*4
Gu Mu†Lin Ling Liu Bo Liu Xiao-Lin Huang Shi-Ming Ni Chen
(Shanghai Key Laboratory of Special Artificial Microstructure Materials and Technology,Department of Physics,Tongji University,Shanghai200092,China)
(Received 5 June 2009;revised manuscript received 17 August 2009)
The electronic structure of M’type GdTaO4is studied by first-principle pseudopotential calculation within the frame of density-functional theory.The calculated band structure of M’-GdTaO4revealed that the top of the valence band is dominated by O-2p and the bottom of the conduction band is dominated by e orbits of Ta-5d.The spin-up and spin-down electrons of Gd-4f are located at 6.27 eV below the top of the valence band and at 3.01 eV above the bottom of the conduction band when on-site Coulomb interaction Ueff=8 eV is applied.The calculated refraction index of M’-GdTaO4is 2.24 which is in good agreement with the result abtained from the Gladstone-Dale relation.
M’type GdTaO4,first-principle calculation,band structure,density of states
book=13,ebook=13
*国家自然科学基金(批准号:50672068,10875085)资助的课题.
†E-mail:mgu@tongji.edu.cn
猜你喜欢
装备维修技术(2021年36期)2021-10-25
北京航空航天大学学报(2021年7期)2021-08-13
弹箭与制导学报(2021年3期)2021-07-30
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
西安交通大学学报(2018年12期)2018-12-12
吉首大学学报(自然科学版)(2018年3期)2018-07-03
物理学报(2017年1期)2017-07-31
导航定位学报(2015年2期)2015-06-05
