补体C3基因敲除小鼠基因型鉴定的PCR条件优化
2010-08-06 11:42:16郑静李丽梅阳泰刘阳刘进李敏惠杨淑霞邹强
成都医学院学报 2010年3期
郑静,李丽梅,阳泰,刘阳,刘进,李敏惠,杨淑霞,邹强
(成都医学院1.科研实验中心,2.药学系2006级药物制剂本科班,四川 成都 610083)
基因敲除小鼠(gene knockout mouse)是利用基因敲除技术使正常实验小鼠的基因组特定位点上目的基因失活,经过繁殖筛选后所得到目的基因表达缺陷小鼠,是研究被敲除的目的基因的理想实验材料[1-3]。为了获得可靠的动物模型,基因敲除小鼠在用于实验研究前,必须进行准确、可靠的基因型鉴定,目前所采用的方法主要有聚合酶链式反应(polymerase chain reaction,PCR)和 Southern杂交。Southern杂交虽然准确性相当高,但是实验步骤比较复杂、工作周期较长,而且花费比较高;PCR法因其具有快速、灵敏、费用低等特点,逐渐取代了传统的Southern杂交,被广泛应用于对基因敲除小鼠基因型的鉴定实验中[4,5]。但在PCR扩增中,如果实验条件不合适,如引物设计不合理、模板DNA质量不高或被污染、反应体系中各成分因子比例失衡、PCR仪温控不准确、人为操作失误等,都容易导致不可靠或无可重复性的PCR扩增结果[6]。
本实验室从美国JAX实验室引进了补体C3基因敲除小鼠,补体C3基因的敲除是通过插入Neo基因后导致补体C3基因失活实现的。在最初对补体C3基因敲除小鼠基因型鉴定实验中,依照常规的引物设计思路,根据插入的Neo基因序列设计PCR引物,但是由于Neo基因是实验室常用的表达克隆载体,我们发现不论在DNA提取过程中还是PCR反应体系配制过程中,都存在模板DNA被外源Neo基因污染的风险,从而造成假阳性的出现。另外,由于PCR扩增反应不充分等因素,经常会出现不清晰的电泳条带,加大了实验结果的不可靠性。所以,本实验室改进了引物设计的方法,针对两个基因(Neo和C3)的序列设计一对引物,有效地避免了因模板DNA的污染而造成的假阳性。同时,本实验室通过对PCR反应各个因素的反复优化,建立了一套有效可靠的PCR反应体系及反应条件,并且当PCR反应的某个因素发生改变时,如改变退火温度、Mg2+浓度、引物浓度及循环次数,在一定的变化范围内,PCR扩增仍能达到理想的效果,同时,通过对不同浓度模板DNA的扩增,检测PCR优化体系的反应灵敏性。
1 材料与方法
1.1 实验材料
1.1.1 实验动物 补体C3基因敲除小鼠(stock number:003641)从美国JAX实验室引进,品系为B6.129S4-C3tmlCrr,雌、雄杂合子小鼠(C3+/-)各6只。
1.1.2 主要试剂 全基因组DNA提取试剂盒购自Promega公司;PCR反应缓冲液(不含Mg2+)、dNTP、Mg2+及Taq DNA聚合酶均购自MBI公司;PCR检测引物由上海生工合成;电泳级琼脂糖购自西班牙BIOSCIENCE公司;DNA标准品D2000购自Solarbio公司。
1.1.3 主要仪器 Thermo Biofuge stratos台式高速冷冻离心机;BioRad PCR扩增仪PTC-200;Bio-Rad电泳及凝胶成像系统。
1.2 实验方法
1.2.1 小鼠全基因组DNA的提取 补体C3基因敲除小鼠共12只,各取外周血120μ l,按照DNA提取试剂盒提取小鼠全基因组DNA。每个样品取10 μ l进行混合,制备成混合模板,电泳鉴定与分光光度计检测提取DNA的质量和纯度,混合模板用于PCR反应,保存于-20℃.
1.2.2 引物合成 引物1:针对插入片段Neo基因上的序列(5'-CGG CAT TCT GCA CGC T TC AA-3');引 物 2:针 对补 体 C3 基 因上 的序 列(5'-GT T CAT TCA GGG CAC CGG ACA-3'),扩增片段为280 bp,由上海生工合成。
1.2.3 PCR反应体系及扩增程序 PCR反应体系为 25 μ l:0.5 μ l dNTP(10 mmol/L),3 μ l MgCl2(25 mmol/L),2.5 μ l 10 ×PCR 缓 冲 液(不 含Mg2+),0.25 μ l引物 1(20 μ mol/L),0.25 μ l引物2(20 μ mol/L),0.2 μ l Taq DNA 聚合酶(5 U/μ l),2 μ l DNA 模板 ,超纯水补至 25 μ l.
PCR扩增程序为:94℃变性3 min;94℃变性20 s,64℃退火30 s,72℃延伸35 s,35个循环;72℃延伸 10 min.取PCR产物10 μ l于 2%琼脂糖凝胶电泳,D2000作为分子量标准。
1.2.4 PCR反应体系的检测 在初步建立的PCR反应体系基础上,分别通过改变Mg2+浓度、引物浓度、退火温度,分析各种因素对PCR结果的影响。在改变PCR反应体系中某因素时,其它条件保持不变。
1.2.5 PCR扩增循环次数的检测 采用20、25、30、35、40、45个循环次数对反应体系进行 PCR扩增。
1.2.6 PCR灵敏性检测 对混合DNA进行两倍稀释,得到不同浓度的模板DNA用于PCR扩增。
2 结果
2.1 Mg2+浓度
本实验设置了 8个 Mg2+浓度梯度,分别为0.5、1、1.5、2、2.5、3、3.5 和 4 mmol/L,结果表明,当Mg2+浓度低于1.5 mmol/L时扩增不出任何结果,Mg2+浓度在2-4 mmol/L的浓度范围内,PCR扩增均能得到较为清晰的条带,而且没有出现非特异性扩增(图1)。
2.2 引物浓度
对引物进行两倍稀释,共设置了8个浓度梯度,分别为 0.003125、0.00625、0.0125 、0.25、0.05、0.1、0.2 和 0.4 μ mol/L.结果显示 ,随着引物浓度的降低,扩增得到的特异性片段量越少,电泳条带亮度越低,当引物浓度低于 0.0125 μ mol/L时,扩增不出特异性条带,而当引物浓度达到0.1 μ mol/L时,得到的引物二聚体逐渐增多,但是在0.0125-0.4 μ mol/L范围内均未出现非特异性条带(图2)。
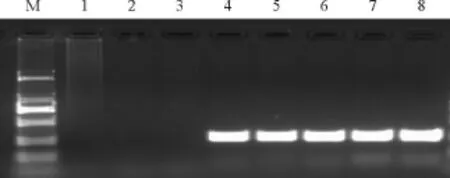
图1 不同Mg2+浓度对PCR结果的影响Fig.1 The products of PCR under different Mg2+concentration
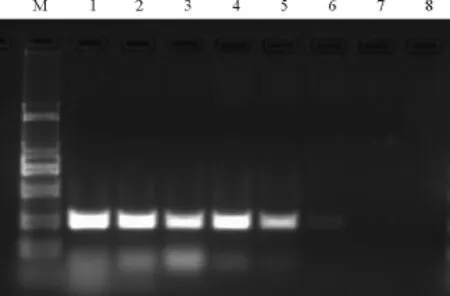
图2 不同引物浓度对PCR结果的影响Fig.2 The products of PCR under different primer concentration
2.3 循环次数
分别采用 20、25、30、35、40、45 个循环进行PCR扩增。结果可见,循环次数在20和25则无法扩增出足够的特异性条带,随着循环次数的增加,电泳条带亮度越高,但并无非特异性带的产生,采用30-45个循环却可以扩增出单一的特异性条带(图3)。
2.4 退火温度对PCR反应的影响
应用PCR仪的温度梯度功能对50℃-70℃的退火温度进行多次筛选,相应的退火温度值分别为50.5、51.5、55.5、58.4 、61.8、64.6、68.4、69.6℃。50.5、51.5℃时泳道均出现非特异性扩增带,55.5℃-69.6℃时泳道的特异性扩增条带非常清晰(图4)。
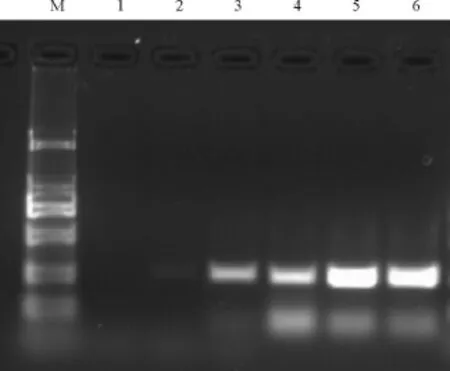
图3 不同循环次数对PCR结果的影响Fig.3 The products of PCR in different cycles
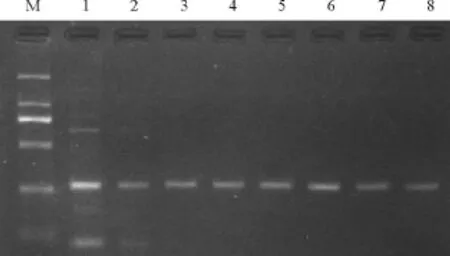
图4 不同退火温度对PCR结果的影响Fig.4 The products of PCR in different annealing temperature
2.5 PCR优化体系灵敏度检测
通过调整体系中模板DNA浓度,检测优化后PCR反应的灵敏性,加入反应体系的DNA体积为2 μ l.ND1000分光光度计的检测结果显示,本次实验所用的模板DNA浓度为90 ng/μ l,对模板DNA进行两倍稀释,得到的浓度分别为 45、22.5、11.25、5.63 、2.81、1.41 和 0.7 ng/μ l.结果可见,DNA 浓度越高,扩增产物的电泳条带越清晰,DNA浓度为0.7 ng/μ l时,无法扩增出任何结果,因此优化后的PCR反应能够检测的最低DNA浓度为1.41 ng/μ l,即DNA的总量约2.82 ng(图5)。
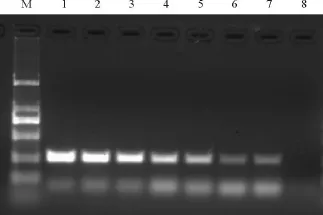
图5 不同DNA浓度对PCR结果的影响Fig.5 The products of PCR in different DNA concentration
3 讨论
PCR技术,是一种体外扩增特异性DNA片段的技术,在反应过程中,目的DNA片段呈指数级增长,可在短时间内获得数百万个特异性DNA序列的拷贝[7]。PCR反应的各个要素,如Mg2+、引物、Taq DNA聚合酶、模板DNA、退火温度、循环次数等都会影响PCR的扩增结果[8]。Mg2+浓度过低,酶的活力降低,浓度过高,则催化生成非特异性条带;引物浓度过低,影响引物与模板的结合,无法获得足够的PCR产物,引物浓度过高,容易由于引物间相互结合产生过多引物二聚体,甚至增大错配的几率,产生非特异性条带;PCR反应的循环次数少,产物量低,无法进行电泳检测,循环次数太多,反应体系中的各种反应试剂消耗完,反应进入平台期之后,则可能会出现非特异性条带。因此,各反应因子的优化组合是获得特异性、可重复性PCR扩增结果的前提,也是扩增反应是否成功的决定因素。另外,模板DNA质量与浓度也是PCR反应成功的一个重要条件,模板DNA量必须保证在PCR能够检测的范围之内,过低量的模板DNA,无法扩增出特异性片段;在提取模板DNA的过程中,除了避免由于实验材料、试剂、人为操作等多方面影响而带入的污染以外,也要考虑到模板DNA之间的交叉污染。
本实验室为了避免由于模板DNA污染引起假阳性结果出现而采取了一些相应的手段。一方面,改进了PCR引物的设计方法;另一方面,本实验室工作人员采用在不同的实验室分别进行DNA提取和配置PCR反应体系的工作,利用空间的分割从最大程度上避免DNA的外源性污染,在DNA提取的过程中采用了过柱的方法,避免了模板DNA之间的交叉性污染。后期的实验证明,本实验室后期采用的这两种方法有效地避免了假阳性现象的出现。
其次,在设计了有效引物之后,本实验室经过一系列优化实验建立了一套稳定可靠的PCR体系,这个PCR体系能够在PCR反应中各个因素发生不同程度的改变时,也能够扩增出单一、清晰的产物条带,得到稳定可靠的鉴定结果。本次实验结果表明,该反应体系Mg2+浓度在2-4 mmol/L,引物浓度在0.025-0.4 μ mol/L,循环次数在 30-45之间均能扩增得到清晰均一的特异性条带;同时,引物退火温度在55.5℃-69.6℃范围内均适用;另外,通过对不同浓度模板DNA的扩增检测了优化后PCR体系的灵敏性,当Taq DNA聚合酶用量为1 U时,能够检测的最低DNA浓度为1.41 ng/μ l,即模板DNA在反应体系中的总量为约2.82 ng.本次实验证明,通过对补体C3基因敲除小鼠基因型鉴定的PCR条件优化,构建一套可靠、容错率高、适用范围广的PCR体系,可以避免由于每次实验操作或仪器温度控制的不稳定性对PCR反应造成的影响,确保每次实验都能够避免非特异性条带的产生,得到清晰、可靠的结果。
目前,PCR法已经成为基因敲除小鼠基因型鉴定最为常用的一种方法,通常根据插入基因片段的序列或者替代基因的序列设计引物,通过PCR扩增检测小鼠基因型。本实验室在以往对基因敲除小鼠基因型鉴定的工作过程中发现,这种方法设计的引物往往会在PCR过程中因为外源性DNA污染而出现假阳性,出现的概率由于各个实验室对污染源的控制严格程度不同而定,但即使在要求严格的实验室中,偶尔出现了假阳性也很难察觉。因此,在进行基因型鉴定过程中,有效避免外源性污染带入的不可靠的实验结果就显得非常重要。本实验利用引进小鼠是通过插入Neo基因后导致补体C3基因失活构建的原理,将引物设计为一条针对插入片段Neo基因上的序列,而另一条针对补体C3基因上的序列,只有当模板DNA满足于既有Neo基因又有补体C3基因并且两个基因相连时,才能进行PCR扩增,这种引物的设计理念区别于根据一个基因来设计引物的普遍观念,大大降低了PCR过程中假阳性的出现,不但为基因敲除动物基因型的鉴定提供了新的实验思路,也能应用于其它转基因物种的鉴定,有助于设计更为适用的引物,构建得到稳定、可靠、重复性高的PCR体系。
总之,PCR实验结果受诸多因素的影响,为了保证能够得到稳定可靠的实验结果,在建立一个新的PCR反应体系或者改变已经形成的一个成熟体系时,对各个因素的优化是非常必要的。除了PCR实验中涉及到的反应体系和反应条件以外,仪器设备、实验试剂、操作手法等因素都可能对PCR扩增的结果产生影响。本实验室通过改进PCR引物的设计方法,反复优化PCR反应因子,得到了一套稳定的PCR检测方法。经过本次实验,证明了这套方法在PCR某因素发生一定改变的情况下,仍能够扩增出单一、稳定、可信的PCR产物,适用于之后大量的补体C3基因敲除小鼠繁育鉴定实验中。同时,实验过程中利用空间的分割避免了DNA的外源性及交叉性污染,进一步提高了PCR结果的可靠性。
[1]Mannstadt M.The knockout mouse--a revolutionizing model in biomedical research[J].M ed klin,1997,92(9):558-560.
[2]Sassi F,Bejaoui M,Ayed K.A congenital deficiency of the C3 fraction of complement.A familial study[J].Tunis Med,2003,81(5):354-358.
[3]Huber-Lang M,Sarma J.V,Zetoune F.S,et al.Generation of C5a in the absence of C3:a new complement activation pathway[J].Nat M ed,2006,12(6):682-687.
[4]曲玉秀,汤家铭,连安,等.纯合子NEOr转基因小鼠品系的建立及鉴定[J].中国实验动物学报,2004,12(2):76-81.
[5]金悠,吴鹏飞,王芳.PICK1基因敲除小鼠的繁育及子代基因型鉴定[J].华中科技大学学报(医学版),2008,37(3):380-383.
[6]Roux KH.Optimization and troubleshooting in PCR[J].PCR Methods Appl.1995,4(5):185-194.
[7]Arnhelm N,Levenson CH.Polymerase Chain Reaction[J].Chem Eng News,1990,68(40):36-47.
[8]段新华,刘诚明.关于PCR扩增体系优化的实验研究[J].现代肿瘤医学,2004,12(4):294-298.
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04 11:40:51
生物学通报(2022年1期)2022-11-22 08:12:18
南京林业大学学报(自然科学版)(2021年5期)2021-10-13 02:06:16
昆明医科大学学报(2021年8期)2021-08-13 09:00:02
昆明医科大学学报(2021年2期)2021-03-29 07:42:24
中国医药指南(2017年3期)2017-11-13 02:55:32
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
广西林业科学(2016年3期)2016-03-16 05:43:25
中国康复理论与实践(2015年10期)2015-12-24 05:42:46