
异源抗体对接方法预测流感病毒血凝素B细胞构象表位
2010-08-06 08:04:56王靖飞吴春艳冯宝龙
中国预防兽医学报 2010年1期
梁 瑾,王靖飞*,吴春艳,冯宝龙
(1.中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室,黑龙江哈尔滨150001;2.东北农业大学继续教育中心,黑龙江哈尔滨150030)
抗体通常能够特异性地识别抗原蛋白质表面的特定构象及空间位置,这些抗原蛋白表面的特定构象及空间位置就代表着抗原的表位,因此表位被定义为处于抗原表面维持这些特殊构象及位置的与抗体上抗原结合位点相互作用的氨基酸残基[1]。表位根据与抗原受体结合的不同可分为B细胞表位和T细胞表位。B细胞表位是抗原中可被B细胞受体或抗体特异性识别并结合的线性片段或空间构象性结构。从空间结构分析,B细胞表位可分为连续性的线性表位和不连续性的空间构象性表位。线性表位,是由肽链上顺序连续的氨基酸组成;构象性表位,是由那些在空间结构上接近,但顺序上不连续的氨基酸组成,具有高度的空间依赖性。表位是蛋白质抗原性的基础,准确的预测B细胞表位不仅有助于基础免疫学研究,而且对疫苗的研究和开发、疾病的预防和诊断也有重要意义。
目前,绝大多数B细胞表位的预测方法都是基于蛋白质的一级或二级结构,但这些方法只能用来预测由连续的氨基酸残基构成的线性表位,而基于蛋白质的三级结构的构象表位预测方法相对较少。分子对接是近几年发展起来的从构象上研究蛋白质-蛋白质相互作用的方法之一,主要用来分析分子间的相互作用模式,以及预测复合物结构[2]。分子对接一般分为刚性对接和柔性对接,刚性对接是基于蛋白质相互作用界面的形状互补,参与对接的分子构象不发生变化,仅改变分子的空间位置与姿态。柔性对接方法在对接过程中允许研究体系的构象发生自由变化。抗体结合抗原的区域大都在互补决定区(CDR)区,CDR区主要与抗原表位发生相互作用,表位只有具有和CDR区结构互补的特征才更有利于与CDR区结合[3]。能否利用刚性分子对接技术来预测B细胞表位是一个值得探索的问题。
本研究选用了PDB数据库中5个非流感病毒的抗体,同已知2个抗原表位的A/Aichi/2/68(H3N2)HA进行分子对接,分析这5个非流感病毒的抗体是否能预测出A/Aichi/2/68(H3N2)HA的2个已知的抗原表位,从而探索这种方法的可行性。
1 材料和方法
1.1 分子对接的受体和配体的准备 受体:从PDB数据库中任意挑选了5个抗原抗体复合物晶体结构中的抗体作为受体,复合物分别为:艾滋病毒包膜糖蛋白GP120-单克隆抗体(MAb)复合物晶体结构(1G9M)、人鼻病毒外壳蛋白-MAb复合物晶体结构(1RVF)、朊病毒蛋白质-MAb复合物(1TPX)、N9亚型流感病毒神经氨酸酶-MAb复合物晶体结构(1NCA)、卵清溶菌酶-MAb复合物晶体结构(1A2Y);配体:选用流感病毒A/Aichi/2/68(H3N2)的HA三聚体结构即1HGF,取出其中的1个HA单体作为配体。
1.2 A/Aichi/2/68(H3N2)已知表位的确定 A/Aichi/2/68(H3N2)的HA抗原抗体复合物的晶体共有6个,分 别 为 1EO8[4]、 1QFU[5]、 1KEN[6]、 2VIT(2VIR 2VIS)[7],这些晶体中抗体实际作用于2个表位:分别是1EO8、1QFU复合物中MAb所结合的表位及1KEN、2VIT(2VIS 2VIR)复合物中MAb所结合的表位。本研究将1EO8及2VIT的表位定义为A/Aichi/2/68(H3N2)的已知表位,用于预测结果的分析,分别定义为Epitope 1和Epitope 2。
1.3 受体和配体的处理 受体:1HGF为三聚体形式,取其单体形式,由于抗原表位在HA1上,为了增加运算速度,只保留HA1,存为1HGF-HA;配体1G9M、1RVF、1TPX、1NCA、1A2Y均为抗原抗体复合物,只保存其抗体,分别命名为1G9M-Ab、1RVF-Ab、1TPX-Ab 、1NCA-Ab、1A2Y-Ab。
1.4 抗体和HA的分子对接 在Discovery studio2.1软件包中的Dock Proteins(ZDOCK)模块中,1G9MAb、1RVF-Ab、1TPX-Ab 、1NCA-Ab、1A2Y-Ab分别与1HGF-HA通过ZDOCK进行分子对接[8],其中抗体作为配体,而1HGF-HA作为受体。对受体和配体中不能参与表位形成的氨基酸做了对接限制。从ZDOCK输出的文件中,所有的Pose都用Refine dock proteins(RDOCK)程序进行优化[9]。
1.5 表位的确定 根据对X射线衍射所确定的抗原抗体复合物推断,有3种对表位定义的方法[10]:(1)当抗体结合到抗原上后,抗原上的任何一个原子所属残基的表面可及性面积与原来相比增加了1埃,这样的残基可以被认为是表位残基;(2)对接复合物中与抗体上任何原子的作用距离小于等于4埃的抗原上的任何原子所在的氨基酸残基被认为是表位残基;(3)对接复合物中与抗体上任何原子的作用距离小于等于5埃的抗原上的任何原子所在的氨基酸残基被认为是表位残基。但这3种不同表位定义的方法并不会造成结果上的差异。在本研究中我们选择第2种确定表位的方法。一些研究者证明阈值设为4埃能够准确的确定92%的表位残基,并且仅有1%的非表位残基被确定为表位残基[11]。
1.6 预测结果分析 结果准确性分析主要通过对作用表面氨基酸的结构以及种类进行统计,前者利用Discovery studio2.1计算均方根偏差(RMSD)来衡量,后者则用预测结果中和实际表位一致的氨基酸数量除以已知表位相应氨基酸的数量获得。
2 结果
2.1 预测表位的空间构象 将1G9M-Ab、1RVFAb、1TPX-Ab、1NCA-Ab、1A2Y-Ab 与 1HGF-HA对接后经过RDOCK优化的对接构象分别同1EO8、2VIT晶体结构重叠,做抗原抗体作用表面氨基酸残基C-α原子的RMSD值,得到抗原抗体作用表面同晶体中的相应表面重叠最好的构象(图1)及其RMSD值(表1)。从RMSD结果分析,所有通过对接方法确定的可能的B细胞表位与已知表位C-α原子之间的RMSD均小于0.6,表明对接结果同晶体结构中抗原抗体相互作用表面的氨基酸残基在空间分布上很接近,即用这5个非流感病毒的抗体均对接到A/Aichi/2/68(H3N2)HA上与2个已知的抗原表位非常接近的氨基酸表面(图2)。
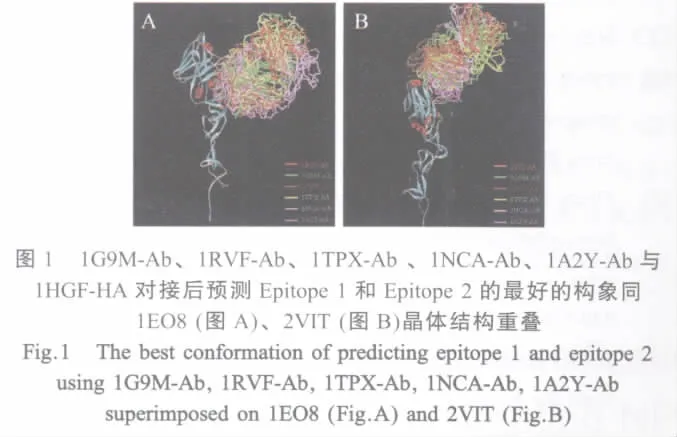
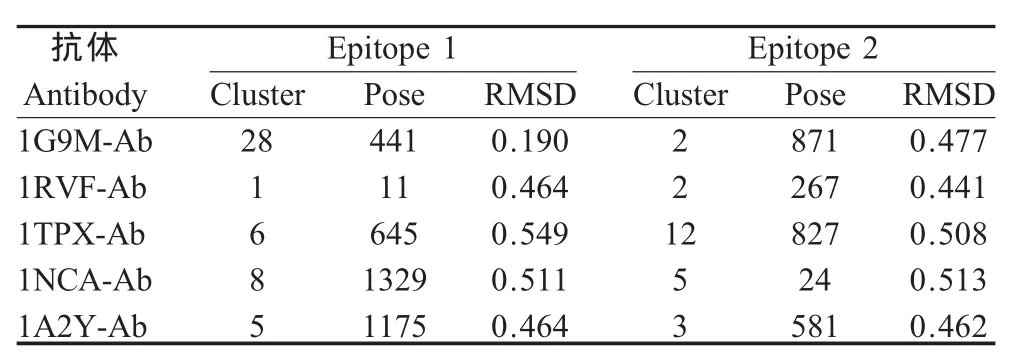
表1 对接构象同1EO82VIT晶体结构重叠后的抗原抗体接触表面氨基酸残基C-α原子的均方根偏差Table 1 The Cα atoms RMSD calculation of interface residue after superimposition

2.2 预测抗原表位的氨基酸组成 A/Aichi/2/68(H3 N2)的 2个已知表位(Epitope 1、Epitope 2)分别为1EO8-Ab和2VIT-Ab抗体结合表位,其氨基酸数量分别为 15和 18。 1G9M-Ab、1RVF-Ab、1TPX-Ab、1NCA-Ab、1A2Y-Ab对A/Aichi/2/68(H3N2)HA的这2个表位预测的准确率在67%~100%之间,平均为77.6%,均高于60%,而且每个抗体均可结合到Epitope 1和Epitope 2。这些结果提示,用1个异源抗体可以对1个抗原蛋白上的不同B细胞表位进行预测;同时,不同的异源抗体也可以对1个抗原蛋白上同1个B细胞表位进行预测(表2、表3)。
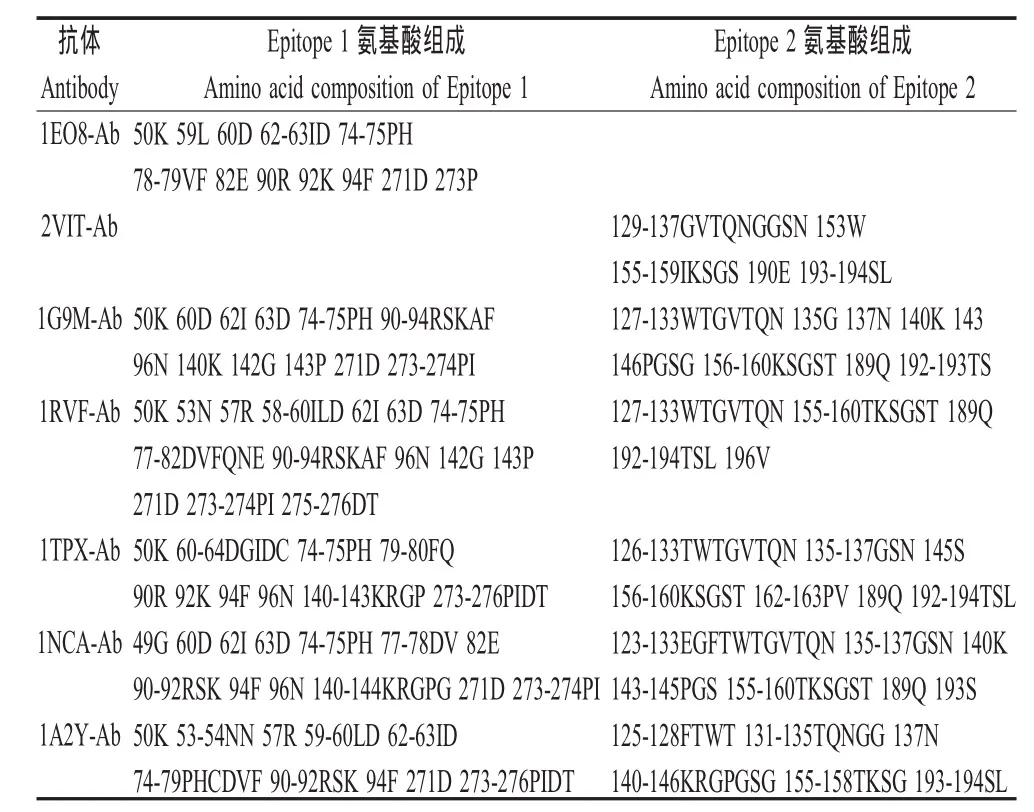
表2 A/Aichi/2/68(H3N2)的HA的表位氨基酸组成及预测表位的氨基酸组成Table 2 The epitope residues of hemagglutinin of strain A/Aichi/2/68(H3N2)and the predicted epitope residues
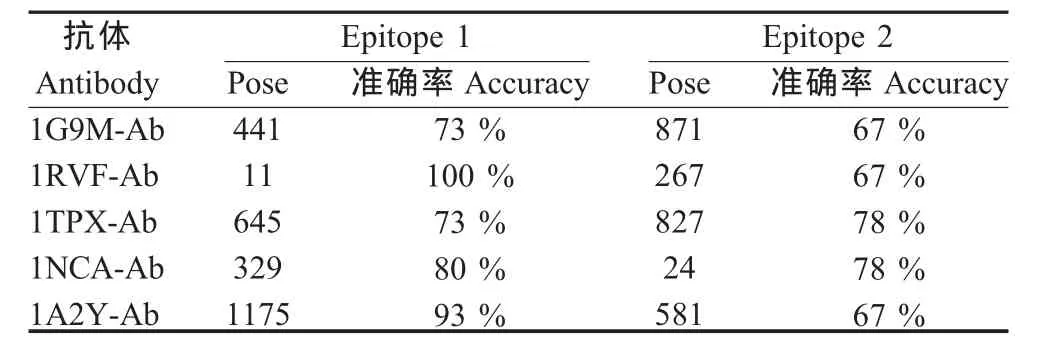
表3 各抗体预测A/Aichi/2/68(H3N2)的HA的表位氨基酸的准确率Table 3 The accuracy of the predicted epitope of the strain A/Aichi/2/68(H3N2)
3 讨论
对预测结果的评估主要从两方面来进行,一方面是抗原抗体作用表面的RMSD值;RMSD值是在结构建模和预测中最为常用的指标,用来测量2个或多个结构数据之间的差异程度,RMSD值越小,则建模或预测结果越可靠,Chen等认为预测构象同原晶体构象重叠之后,其蛋白质与蛋白质相互作用表面C-α原子的RMSD如果小于2.5埃,则可认为其预测结果是正确的[12],从结果中可以看出使用非流感病毒抗体同流感病毒HA进行分子对接,最好的对接结果与原晶体结构重叠后抗原抗体作用表面氨基酸C-α原子的RMSD是小于2.5埃的,符合这个评判标准。另一方面是组成抗原表位的氨基酸残基预测的准确性;Kulkarni-Kale等认为,如果大于等于60%的抗体结合位点的残基被预测准确,那么这种预测结果就能被认为是正确的[13]。从结果中可以看出使用非流感病毒抗体同流感病毒HA进行分子对接,最好的对接结果的表位氨基酸残基预测的准确性均大于60%,符合这个评判标准。
结果显示,使用非流感病毒抗体能够比较准确的预测流感病毒HA的抗原表位。但是如果我们在实际中应用这种方法,则会面临一些问题:首先,最好的构象并不一定在最大的聚类中,比如以1TPX-Ab、1NCA-Ab、1A2Y-Ab为配体的最好的对接构象均不在最大的Cluster中,即使是在最大的Cluster中如以1RVF-Ab为配体的最好的对接构象也不是得分最高的Pose这将对我们筛选出能够正确预测表位的构象造成一定的困难。其次,对接构象所预测的抗原表位残基的数目均多于实际抗原表位残基,这是由对接过程中抗原抗体的作用距离来决定的,由于仅仅根据形状互补做刚性对接,限制条件比较少,因此抗原抗体的作用距离会比较近,则与抗体作用距离为4埃的抗原上的氨基酸残基的数目就多,从而预测的抗原表位氨基酸残基中会有一部分并不属于真正的抗原表位,如何将这部分氨基酸筛去也是需要解决的问题。因此,如果将这种方法应用于实际中抗原表位的预测,还需增加筛选条件,来解决以上问题,从而提高预测的准确性。
由于目前预测表位方法的准确性均不高于40%[14],我们期望找到一种能够比较准确的预测表位的方法。本研究即通过刚性分子对接的方法,从构象上探测仅仅根据抗原抗体表面的形状互补是否能准确预测抗原表位。结果表明,这种方法是可行的,并且最好的结果预测表位氨基酸的准确率高于60%。这一结果对以后建立一套完整的异源抗体预测抗原表位的方法具有一定的参考价值,并对正确而详细地绘制抗原表位图谱有着一定的指导意义。
[1]Van Regenmortel M H.From absolute to exquisite specificity reflections on the fuzzy nature of species,specificity and antigenic sites[J].J Immunol Methods,1998,216,37-48.
[2]李春华,马晓慧,陈慰祖,等.蛋白质-蛋白质分子对接方法研究进展[J].生物化学与生物物理进展,2006,33(7):616-621.
[3]Rubinstein N D,Mayrose I,Halperin D,et al.Computational characterization of B-cell epitopes[J].Mol Immunol,2008,45(12):3477-3489.
[4]Fleury D,Daniels R S,Skehel J J,et al.Structural evidence for recognition of asingle epitope by two distinct antibodies[J].Proteins,2000,40(4):572-578.
[5]Fleury D,Barrère B,Bizebard T,et al.A complex of influenza hemagglutinin with aneutralizing antibody that binds outside the virus receptor binding Site[J].Nat Struct Biol,1999,6(6):530-534.
[6]Barbey-Martin C,Gigant B,Bizebard T,An antibody that prevents the hemagglutinin low pH fusogenic transition[J].Virology,2002,294(1):70-74.
[7]Fleury D,Wharton S A,Skehel J J,et al.Antigen distortion allows influenza virus to escape neutralization[J].Nat Struct Biol,1998,5(2):119-123.
[8]Chen R,Weng Z.A novel shape complementarity scoring function for protein-protein docking[J].Proteins,2003,51:397-408.
[9]Li L,Chen R,Weng Z.RDOCK:Refinement of rigid-body protein docking predictions[J].Proteins,2003,53:693-707.
[10]Blythe M,Flower D.Benchmarking B cell epitope prediction:underperformance of existing methods[J].Protein Sci,2005,14:246-248.
[11]Haste A P,Nielsen M,Lund O.Prediction of residues in discontinuous B-cell epitopes using protein 3D structures[J].Protein Sci,2006,15(11):2558-2567.
[12]Chen R,Li L,Weng Z P.ZDOCK:An initial-stage protein-docking algorithm[J].Proteins,2003,52:82-87.
[13]Kulkarni-Kale U,Bhosle S,Kolaskar A S.CEP:a conformational epitope prediction server[J].Nucleic Acids Res,2005,33(Web Server issue):168-171.
[14]Ponomarenko J V,Bourne P E.Antibody-protein interactions:benchmark datasets and prediction tools evaluation[J].BMC Struct Biol,2007,7:64.
猜你喜欢
遵义医科大学学报(2023年4期)2023-05-05 05:05:16
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
中国免疫学杂志(2017年1期)2017-01-17 04:53:25
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
畜牧兽医学报(2015年3期)2015-07-05 08:22:53
医学研究杂志(2015年6期)2015-07-01 17:41:11
应用化工(2014年7期)2014-08-09 09:20:23
