
中国沿海长蛸群体16S rRNA基因的遗传变异研究
2010-07-12 08:18吕振明常抗美迟长凤吴常文
浙江海洋大学学报(自然科学版) 2010年4期
李 焕,吕振明,常抗美,迟长凤,吴常文
(1.浙江海洋学院海洋科学学院,浙江省海洋养殖装备与工程技术重点实验室,浙江舟山 316004)
长蛸Octopus variabilis是我国沿海重要的海洋头足类,又名马蛸、长腿蛸、大蛸,属软体动物门(Mollusca)头足纲(Cephalopada)蛸科(Octopodidae)蛸属动物,分布于我国黄海、渤海、东海和南海海域。其产品肉味鲜美,营养丰富,具有补气养血,收敛生肌的作用,经济价值高,是畅销国际国内市场的水产品[1],也一直是我国重要的海洋捕捞种类之一。但近年来,由于我国沿海头足类资源的大力开发,捕捞强度日益加大,其资源量也日趋衰退[2]。为了更有效地保护和管理我国的长蛸资源,维持其可持续采捕,同时也为今后长蛸资源更合理的人工开发,也有必要对其种群结构和遗传变异作深入的研究,从而为我国长蛸资源的保护和管理提供必要的指导。
线粒体DNA(mtDNA)具有较高的突变率,突变固定后形成的多态性位点可反映出群体遗传特征、种群分化和种属关系的特征,已成为研究动物起源进化、群体遗传、系统发育等的重要标记[3],迄今为止已在枪乌贼[4]、柔鱼[5]、章鱼[6,7]等主要头足类种类中得到广泛研究。本文采用线粒体16S rRNA基因测序技术对我国沿海4个地理群体长蛸的遗传变异和种群结构进行研究,以期为长蛸种质资源的管理和保护提供理论依据,也为长蛸种质评价和优异种质筛选及进一步的良种选育奠定基础。
1 材料与方法
1.1 实验材料
长蛸样品取自中国渤海、黄海、东海海域,采集地点分别为辽宁大连、山东青岛、浙江舟山、福建厦门,各海域取长蛸样本10~12尾,样本取新鲜肌肉保存于95%酒精,带回实验室备用。
1.2 实验方法
1.2.1 基因组 DNA 的提取
参照《分子克隆实验指南》[8],采用常规酚-氯仿法提取基因组DNA,双蒸水溶解后置于-20℃保存备用。
1.2.2 16 S rRNA 基因扩增和测序
16S rRNA基因序列的扩增参照LIN等[9]的方法进行,稍加改进。具体方法为:扩增引物16S f:5’-CGCCTGTTTAHYAAAAACAT-3’;16S r:5’-CCGGTCTGAACTCAGMTCAYGT-3’,引物由上海英骏生物技术有限公司合成;PCR 反应体系总体积为 50 μL,其中含模板 DNA 50~100 ng,2.0 mM MgCl2,0.2 mM 每种dNTPs,0.2 μM 每个引物,3 U Taq酶及 1x缓冲液;反应程序为:94 ℃预变性 5 min后,94 ℃变性 1 min,51℃退火1 min,72℃延伸1 min,共35个循环,最后72℃延伸5 min。PCR扩增产物经1.5%琼脂糖凝胶电泳检测,凝胶成像系统下观察并拍照记录。PCR产物纯化后送至Invitrogen公司测序。
1.2.3 数据分析
DNA序列采用Clustal X 1.83[10]软件进行编辑、校对和排序;采用DNAsp4.10[11]软件对多态位点数、单倍型数、转换与颠换数、核苷酸多样性(π)、单倍型多样性(H)、平均核苷酸差异数(K)等遗传多样性参数进行计算;采用Mega3.1[12]软件进行遗传距离计算和聚类分析,系统树采用UPGMA模型构建,并采用bootstrap(重复次数1 000)检验聚类树各分支置信度;此外,用Arlequin3.01[13]软件中的分子变异分析(AMOVA)方法估算遗传变异在群体内和群体间的分布,并计算群体间遗传分化系数(F-statistics,Fst)及其显著性(重复次数1 000),群体间基因流Nm由公式Nm=(1-Fst)/2 Fst计算而得。采用Tajima’s D检验和Fu’s Fs检验来检测中性假说是否成立,Tajima’s D和Fu’s Fs中性检验结果如为负值并且显著偏离中性,则可能是由于群体扩张引起的[14]。
2 结果
2.1 16S rRNA基因的序列组成及变异
采用Clustal w1.83软件对我国4个长蛸群体共45个个体的16S rRNA序列编辑和比对后,得到512 bp 的同源序列。经 Mega 3.1 软件分析,该序列的 A、T、C、G 碱基的平均含量为 35.2%、39.2%、7.9%、17.7%。A+T的含量(74.4%)明显高于 G+C 的含量(25.6%)。DNAsp4.10软件分析显示,45个个体共检测到48个变异位点,占分析位点总数的9.4%,其中单突变位点3个,简约信息位点45个。同时还检测到32个插入或缺失位点,碱基插入主要发生在234~255 bp之间的区域,以TA重复片段的形式进行,但仅在大连、青岛和舟山3个群体中检测到。多态性遗传参数统计显示,45个个体共检出30个单倍型(表1),其中大连群体11个,青岛群体11个,舟山群体9个,厦门群体2个;总群体单倍型多样性指数(H)为0.964 6,核苷酸多样性指数(Pi)0.032 9,平均核苷酸差异数(K)15.800,显示出较丰富的遗传多样性。
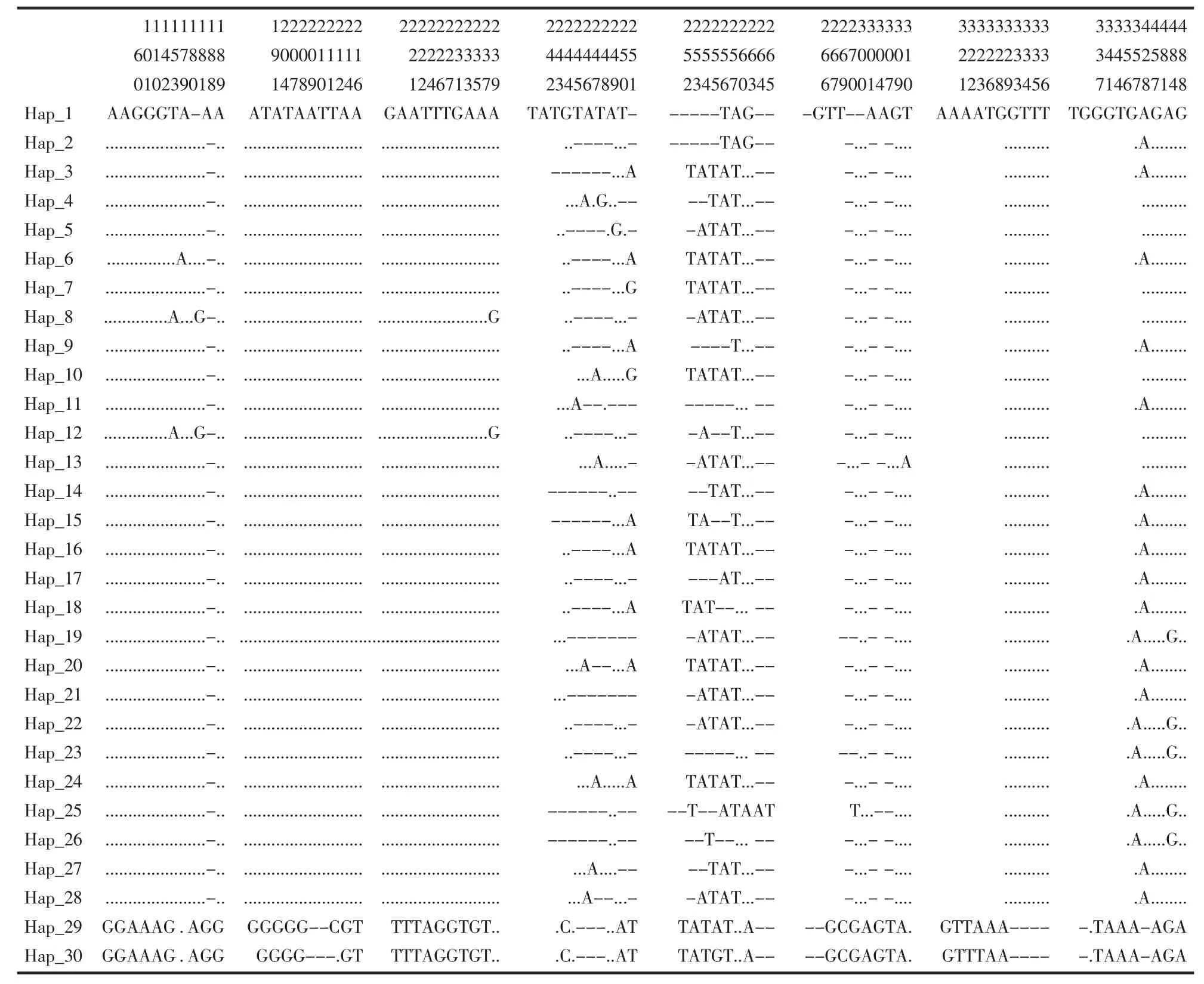
表1 长蛸16S rRNA基因序列单倍型Tab.1 Haplotypes of 16S rRNA in four populations of Octopus variabilis
2.2 长蛸群体的遗传结构分析
对4个群体的16S rRNA基因序列进行比较,结果表明,各群体无论在核苷酸组成上还是在遗传参数上均存在着显著差异。在核苷酸组成上,大连、青岛、舟山3群体核苷酸组成较为相似,但它们与厦门群体却存在43个固定核苷酸的差异,见表1。遗传参数统计表明,各群体多态位点比例在0.586%~0.977%之间;单倍型多样性指数在0.455~0.697之间;核苷酸多样性指数在0.002 0~0.002 8 之间;平均核苷酸差异数在 0.970~1.400 之间,见表2。对4个群体间的遗传分化系数和基因流进行计算,结果见表3,4个群体已产生较大程度的分化,特别是厦门群体与其它3个群体之间分化系数达到0.976 1~0.9799之间,均达到显著水平(p﹤0.01),基因流 Nm 也均小于1;舟山与大连、青岛群体之间虽分化显著(p<0.01),但仍具有一定的基因流(Nm﹥1)。大连与青岛群体之间遗传分化不显著(Fst p﹥0.05)。AMOVA 进一步分析显示在整个遗传变异中有82.07%存在于群体内部,17.93%存在于群体之间,进一步证实了长蛸群体间的分化。
2.3 聚类分析和种群动态
采用Mega 3.1软件对上述4个群体进行UPGMA聚类分析(图1),结果表明,所有个体可以明显聚为2支,一支由大连、青岛和舟山3群体的个体组成,另一支由厦门群体所有个体组成;遗传距离分析表明,厦门群体与其它3个群体的净遗传距离为0.093,而3个群体内部的净遗传距离为0;对4个群体进行Tajima’s D和Fu’s Fs检验,结果表明,尽管有些群体的Tajima’s D和Fu’s Fs值为负值,但均未达到显著水平(p﹥0.05),因此不支持这些群体历史上经过种群扩张。

表2 长蛸群体的遗传变异参数统计Tab.2 Statistics of genetic variation parameter in O.variabilis populations
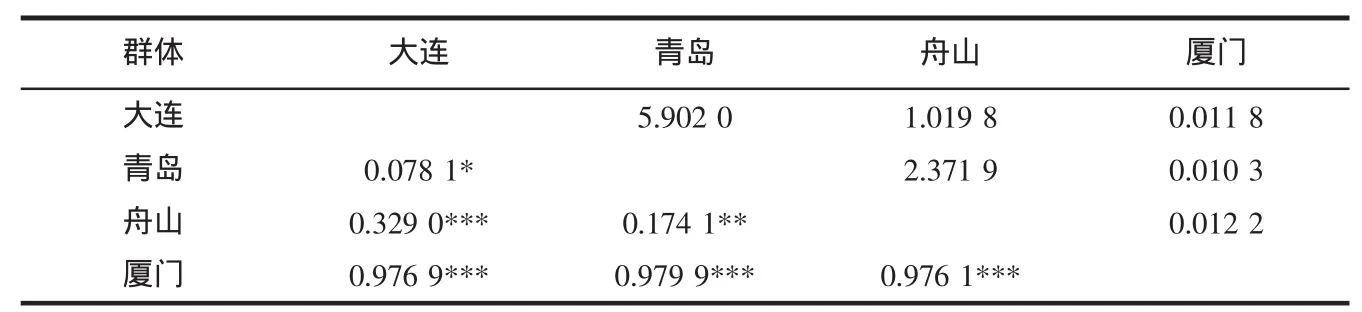
表3 长蛸群体间的遗传分化系数(对角线下)和基因流(对角线上)Tab.3 Genetic fixation index(below diagonal)and gene flow(above diagonal)among Octopus variabilis populations
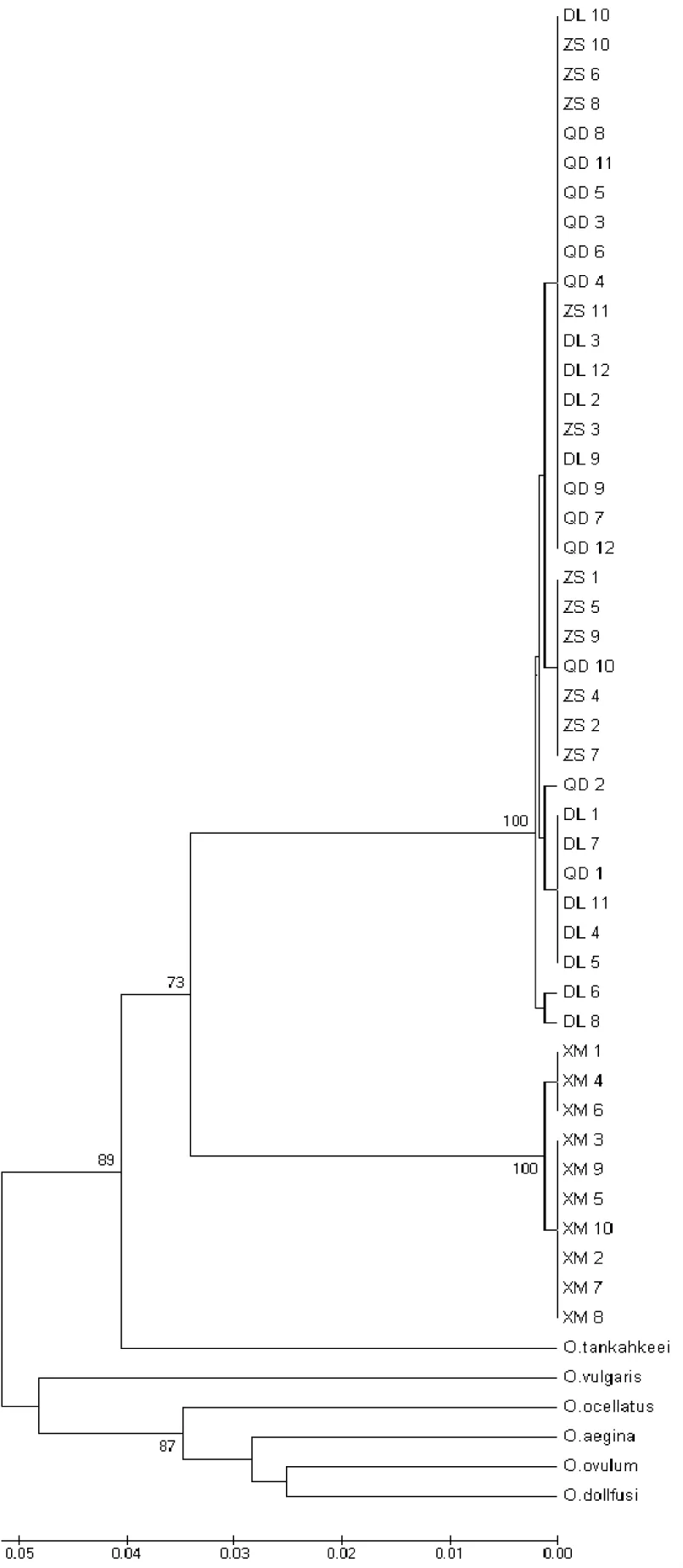
图1 长蛸不同个体的UPGMA聚类分析图Fig.1 UPGMA tree constructed from genetic distance among O.variabilis individuals
3 讨论
遗传多样性不仅是形成生物多样性的基础,也是物种进化潜能的保证。遗传多样性的降低或丧失,对于生活在多变环境中的野生群体是一个极大的威胁[15]。衡量群体DNA遗传多样性的重要指标有单倍型多样性(H)与核苷酸多样性(π)[16]。本文线粒体16S rRNA序列测定的45个长蛸样本中共获得了30个单倍型,多样性指数H达0.964 6,总体核苷酸多样性指数π为0.032 9,表现出非常丰富的遗传多样性,比已研究的其他头足类如真蛸Octopus vulgaris[17]、鱿鱼Thysanoteuthis rhombus[18]、曼氏无针乌贼 Sepiella maindroni[19]、鹦鹉螺Nautilus pompilius[20]等种类要高,表明我国长蛸群体具有较高的进化潜能,为长蛸资源的保护和开发利用奠定了良好的基础。
我国的长蛸群体不仅存在较高的遗传多样性,还存在显著的遗传结构。AMOVA分析是用来测量群体之间遗传分化的重要指标[21],AMOVA分析表明,我国长蛸群体间的遗传变异有82.07%存在于群体间,而仅有17.93%存在于群体内,揭示长蛸不同群体已经形成显著的遗传分化,并具有明显的种群遗传结构。UPGMA聚类分析进一步证实了以上结果,聚类分析表明,我国沿海的4个长蛸群体可以明显地分化为2大类群,一个类群由大连、青岛和舟山3个群体组成,另一个类群由厦门群体组成。两类群间遗传分化系数(Fst)达到0.905 9,并达到极显著水平,基因流仅0.051 9。一般认为,群体之间的Nm<1,说明群体可能由于遗传漂变而发生了分化;而Nm﹥1,表明群体间的基因流水平较高,群体间遗传分化较小;当Nm>4时,种群间的基因交流就更为充分,遗传分化更小[14]。本研究中两类群的基因流远远小于1,说明已经形成了显著的遗传分化。而两类群内部分化则相对较小,大连、青岛和舟山3群体间的的遗传分化系数仅0.078 1~0.329 0,Nm值均大于1,说明它们间仍有较广泛的基因交流,而这3个群体与厦门群体则分化较大。为更好地展示两类群间的遗传分化程度,引入同属的短蛸O.ocellatus、真蛸、砂蛸O.aegina、嘉庚蛸O.tankahkeei、卵蛸O.ovulum及弯斑蛸O.dollfusi作为外群与所有长蛸群体一起作聚类分析,结果发现4个长蛸群体与这些外群种之间遗传差异更为明显,因此认为长蛸两类群的分化仍属种内水平的分化。对两类群的遗传距离进行分析,结果表明两者的净遗传距离已达到0.093;王家玉[22]利用遗传距离D值对物种的不同分类单位间的遗传分异水平作过定量性估计,并指出种群间遗传距离D值的范围是0-0.05,亚种间是0.02-0.2,厦门群体与其它3个群体间的遗传距离均在0.09以上,综合遗传距离、遗传分化系数和基因流数据(Fst>0.97,Nm≤0.02)3个指标来看,厦门群体与其它3个群体间有着显著的遗传隔离,可能已形成亚种水平的分化。至于群体间分化的原因目前还不清楚,但有研究认为许多因素可以造成海洋生物的分化,例如物种的生活史、海域水文条件以及地理历史因素等[24-27]。长蛸主要营底栖穴居生活,仅在繁殖期间有短距离的洄游移动[1],这种特殊生活习性可能使我国长蛸资源各地理群体间不能进行很好的基因交流,最终导致了地理群体的分化;地理历史因素方面,晚更新世是中国陆架海陆频繁变迁的一个重要时期[27],在此期间经历了一系列冰期-间冰期的变化[28],冰期导致海平面下降,亚洲大陆与其附近岛屿之间形成陆桥。尽管陆桥的形成有利于陆生生物的扩散,但是却可以阻碍海洋生物间的交流,从而有助于海洋生物的异域分化[29]。该时期是否造成我国长蛸群体的分化还须结合更多地理学和生物学方面的资料才能证实。
[1]董正之.中国动物志.软体动物门,头足纲[M].北京:科学出版社,1988.
[2]许星鸿,阎斌伦,郑家声,等.长蛸生殖系统的形态学与组织学观察[J].动物学杂志,2008,43(4):77-84.
[3]袁希平,严 莉,徐树英,等.长江流域铜鱼和圆口铜鱼的遗传多样性[J].中国水产科学,2008,15(3):377-385.
[4]FRANK E A.Phylogenetic relationships among loliginid squids (Cephalopoda:Myopsida)based on analyses of multiple data sets[J].Zoological Journal of the Linnean Society,2000,130:603-633.
[5]CARLIN D B,KUNKLE L K,VECCHIONE M.A molecular systematic evaluation of the squid genus Illex(Cephalopoda:Ommastrephidae)in the North Atlantic Ocean and Mediterranean Sea[J].Molecular Phylogenetics and Evolution,2006:1-7.
[6]WARNKE K,SÖLLER R,BLOHM D,et al.A new look at geographic and phylogenetic relationships within the species group surrounding Octopus vulgaris(Mollusca,Cephalopoda):indications of very wide distribution from mitochondrial DNA sequences[J].Zool Syst Evol Research,2004,42:306-312.
[7]PIERTNEY S B,HUDELOT C,HOCHBERG F G,et al.Collins.Phylogenetic relationships among cirrate octopods(Mollusca:Cephalopoda)resolved using mitochondrial 16S ribosomal DNA sequences[J].Molecular Phylogenetics and Evolution,2003,27:348-353.
[8]SAMBROOK J,FRITSCH E F,MANIATIS T.Molecular Cloning:A Laboratory Manual[M].2nd edn.Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,1989.
[9]LIN Xiang-zhi,ZHENG Xiao-dong,XIAO Shu.Phylogeny of the cuttlefishes(Mollusca:Cephalopoda)based on mitochondrial COI and 16S rRNA gene sequence data[J].Acta Oceanologica Sinica,2004,23(4):699-708.
[10]THOMPSON J D,GIBSON T J,PLEWNIAK F,et al.The Clustal X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Res,1997,25:4 876-4 882.
[11]ROZAS J,SANCHE-DELBARRIO J C,MESSENGUER X,et al.DNA polymorphism analyses by the coalescent and other methods[J].Bioinformatics,2003,19:2 496-2 497.
[12]KUMAR S,TAMURA K,NEI M.MEGA3:Integrated software for molecular evolutionary genetics analysis and sequence alignment[J].Briefings in Bioinformatics,2004,5:150-163.
[13]EXCOFFIER L,LAVAL G,SCHNEIDER S.Arlequin ver.3.01:An integrated software package for population genetics data analysis[J].Evolutionary Bioinformatics Online,2005,1:47-50.
[14]吴常文,许逸天,吕振明,等.基于D-LOOP基因的中国沿海鳓鱼(Ilisha elongata)种群遗传结构研究 [J].海洋与湖沼,2009,40(3):330-337.
[15]杨金权,胡雪莲,唐文乔,等.长江口邻近水域刀鲚的线粒体控制区序列变异与遗传多样性[J].动物学杂志,2008,43(1):8-15.
[16]KATUGIN O N.Genetic variation in the squid Berryteuthis magister(Breey,1913)(Oegopsida:Gonatidae)[M]//OKUTANI T,O’DOR R K,KUBODERA T,eds.Recent advances in fisheries biology.Tokyo:Tokai University Press,1993.
[17]TESKE P R,OOSTHUIZEN A,PAPADOPOULOS I,et al.Phylogeographic structure of Octopus vulgaris in South Africa revisited:identification of a second lineage near Durban harbour[J].Mar Biol,2007,151:2 119-2 122.
[18]KITAURA J,YAMAMOTO G,NISHIDA M.Genetic variation in populations of the diamond-shaped squid Thysanoteuthis rhombus as examined by mitochondrial DNA sequence analysis[J].Fisheries Science,1998,64(4):538-542.
[19]ZHENG X D,WANG R C,WANG X F,et al.Genetic variation in population of the common Chinese cuttlefish Sepiella maindroni(Mollusca:Cephalopoda)using allozymes and mitochondrial DNA sequence analysis[J].Shellfish Res,2001,20:1 159-1 165.
[20]SINCLAIR W,BRISKEY L,AAPDEN W,et al.Genetic diversity of isolated populations of Nautilus pompilius(Mollusca,Cephalopoda)in the Great Barrier Reef and Coral sea[J].Reviews in Fish Biology and Fisheries,2007,17:223-235.
[21]张东亚,汪登强,刘绍平,等.怒江濒危鱼类缺须盆唇鱼基于线粒体Cyt b序列的群体遗传结构分析[J].中国水产科学,2009,16(4):477-486.
[22]王家玉译.分子群体遗传学与进化论[M].北京:农业出版社,1975,121-133.
[23]RIGINOS C,VICTOR B C.Larval spatial distributions and other early life history characteristics predict genetic differentiation in eastern Pacific blennioid fishes[J].Proc R Soc Lond B,2001,268:1 931-1 936.
[24]MUSS A,ROBERTSON D R,STEPIEN C A,et al.Phylogeography of Ophioblennius:the role of ocean currents and geography in reef fish evolution[J].Evolution,2001,55:561-572.
[25]RUZZANTE D E,TAGGARL C T,COOK D A.Nuclear DNA basis for shelf-and bank-scale population structure in northwest Atlantic cod(Gadus morhua):Labrador to Georges bank[J].Molecular ecology,1998,7:1 663-1 680.
[26]LIU Y,LIU R L,YE L C,et al.Genetic differentiation between populations of swimming crab Portunus trituberculatus along the coastal waters of the East China Sea[J].Hydrobiologia,2009,618:125-137.
[27]张军强,唐璐璐,邹 昊.晚更新世以来古气候与海平面变化在东海地区的响应[J].海洋湖沼通报,2008(1):25-31.
[28]IMBRIE J,BOYLE E A,CLEMENS S C,et al.On the structure and origin of major glaciation cycles.I.Linear responses to Milankovich forcing[J].Paleoceanography,1992,7:701-738.
[29]刘进贤,高天翔,吴世芳,等.梭鱼的分子系统地理学研究—晚更新世西北太平洋边缘海隔离分化及其有限的扩散能力[J].中国海洋大学学报:自然科学版,2007,37(6):931.
猜你喜欢
今日农业(2022年15期)2022-09-20
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
清华金融评论(2022年4期)2022-04-13
湖南电力(2021年1期)2021-04-13
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
中国饲料(2019年19期)2019-03-25
红土地(2018年7期)2018-09-26
