
复方生脉散的毛细管电泳指纹图谱研究
2010-05-26 07:57季一兵范晓梅朱丹妮
中成药 2010年8期
季一兵, 范晓梅,2, 朱丹妮
(1.中国药科大学,江苏南京210009;2.广东华南新药创制中心,广东广州 510663)
复方生脉散的毛细管电泳指纹图谱研究
季一兵1, 范晓梅1,2, 朱丹妮1
(1.中国药科大学,江苏南京210009;2.广东华南新药创制中心,广东广州 510663)
生脉散;毛细管电泳;指纹图谱
目的:利用毛细管电泳(CE)方法建立中药复方生脉散(红参、麦冬、五味子)的指纹图谱。方法:采用序贯式均匀设计的方法优化CE分离条件,确定生脉散指纹图谱的电泳条件为:以pH为9.5、44 mmol/L硼砂、34 mmol/L SDS为缓冲溶液体系,运行电压25 kV,温度25℃,压力进样50 mbar×100 s,检测波长200 nm。结果:11批生脉散CE指纹图谱中确认了20个主要的共有指纹峰,其中4个峰来自红参,6个峰来自麦冬,13个峰来自五味子,3个峰为红参和麦冬共有,1个峰为麦冬和五味子共有,另外1个为新成分。结论:该方法准确、可靠,可为生脉散的质量评价与控制提供科学依据。
R284.1
A
1001-1528(2010)08-1277-05
生脉散来源于《医学启源》,是已有820年历史的著名古方。生脉散是由红参、麦冬、五味子3味药组成,具有益气生津、养阴复脉的功效,现代临床上被广泛用于治疗心血管疾病,疗效显著[1]。国内外对生脉散的研究主要集中在药理药效[2-3]、临床应用[4-5]和化学成分[6-8]等方面,而对其进行质量评价与控制的研究较少。为了确保生脉散临床用药的有效性和安全性,非常有必要建立指纹图谱来控制其质量。
生脉散的成分较为复杂,而且民间常用水煎煮的方法来治疗疾病,提取液中含有大量水溶性好的极性组分。毛细管电泳法以其柱效高、峰容量大、分析速度快、运行费用低、分析时间短、尤其适合极性大的组分分离分析的特点,在中药分析特别是中药复方分析中的应用越来越广泛。本试验采用毛细管电泳方法对中药复方生脉散的指纹图谱进行研究,尽可能较为全面地反映其整体的化学成分,为生脉散的质量评价与控制提供依据。
1 仪器与试药
1.1 仪器
HP3DCE毛细管电泳仪(Agilent科技有限公司),二极管阵列检测器,Chemstation工作站;未涂层石英毛细管柱(内径50 μm,有效长度57 cm,河北永年光导纤维厂);pHs-25型pH计(上海精密科学仪器有限公司);电子分析天平(美国Sartorius公司)。
1.2 试药
红参、麦冬、五味子药材为市售品,经中国药科大学中药资源研究室秦民坚教授鉴定,分别为五加科植物人参Panax ginseng C.A.Mey.的栽培品经蒸制后的干燥根及根茎;百合科植物麦冬Ophiopogon japonicus(Thunb.)Ker-Gawl.的干燥块根;木兰科植物五味子Schisandra chinensis(Turcz.)Baill.的干燥成熟果实。五味子醇甲(中国药品生物制品检定所,批号 110857-200709)、五味子乙素对照品(批号 110765-200609)。硼砂和SDS为分析纯,甲醇为色谱纯,纯净水(乐百氏)。
2 实验方法
2.1 对照品溶液的制备
取五味子醇甲、五味子乙素对照品适量,精密称定,加甲醇制成每1 mL分别含五味子醇甲、五味子乙素1.0 mg的混合溶液,作为对照品溶液。
2.2 供试品溶液的制备
按照红参-麦冬-五味子=1∶3∶1.5的比例称取药材110 g,加水煎煮3次(各10倍,8倍,6倍量水),每次1 h,合并煎液,滤过,滤液减压浓缩至浓度为1.1 g/mL生药量的浓缩液。取0.5 mL浓缩液加水稀释至10 mL,离心(3 000 r/min)5 min,上清液用0.45 μm微孔滤膜过滤,滤液作为供试品溶液。红参、麦冬、五味子溶液及各味药材阴性对照溶液同法制备。
2.3 毛细管电泳实验条件
通过实验设计方法优化CE分离条件,确定的实验条件如下:以pH为9.5、44 mmol/L硼砂、34 mmol/L SDS为缓冲溶液体系,分离电压25 kV,温度25℃,压力进样50 mbar×100 s,检测波长200 nm。毛细管第一次使用前依次用1 mol/L NaOH、0.1 mol/L NaOH、去离子水冲30 min。每次实验前,用0.1 mol/L NaOH、去离子水各冲洗10 min,缓冲溶液冲5 min。两次运行之间用缓冲溶液冲洗3 min。
3 毛细管电泳条件的选择和优化
3.1 检测波长的选择
采用二极管阵列检测器,结合生脉散中所含化学成分的光谱特征,考察了200、210、254、285 nm的谱图特征。在200 nm波长处出峰最多且峰强度大,一些有末端吸收的有效成分也可被检测。
3.2 主要因素的筛选试验与结果
在预试验中,硼砂浓度、SDS浓度、缓冲溶液的pH值、甲醇比例、温度、电压影响生脉散组分的分离。然而不同实验因素对CE分离的影响程度存在差异,采用正交设计方法从上述六个因素中筛选出显著影响分离的主要因素。选用L18(36)表安排实验,以所得电泳谱图的峰数目作为衡量CE分离好坏的指标。实验结果经方差分析得出,硼砂浓度、SDS浓度两个因素显著影响谱图的峰数目和分离效果。其余的因素则可固定在较优的实验水平,缓冲溶液pH、温度、电压和甲醇比例分别为9.5、25℃、25 kV和0%。
3.3 序贯式均匀设计试验与结果
采用序贯式均匀设计对硼砂和SDS浓度作进一步地优化,选取U7(72)均匀设计表安排实验,每个因素考察7水平。根据最大峰数目来确定最优分离条件,并以此实验条件为中心,缩小实验范围和进行新一轮的优化试验;再以该轮实验中最优条件为中心,继续安排新一轮优化试验,至获得满意的实验结果为止。本文通过三轮均匀设计试验(表1),得到了最佳的分离条件为44 mmol/L硼砂、34 mmol/L SDS,其他同上述条件。在该条件下所得谱图峰的数目最多、分离佳且分析时间适宜。

表1 3轮均匀设计的实验范围及结果
4 方法学考察
4.1 精密度试验
取同一供试品溶液,连续进样6次,共有指纹峰的相对迁移时间RSD<1.6%,相对峰面积RSD<3.4%。
4.2 重复性试验
取同一批样品6份,按2.2项下方法分别制备供试品溶液,在毛细管电泳仪上进样分析。共有峰的相对迁移时间RSD<2.7%,相对峰面积RSD<4.8%,表明该方法具有较好的重复性。
4.3 稳定性试验
取新制备的供试品溶液于3 d内每隔12 h测定一次,共有峰的相对迁移时间RSD<3.2%,相对峰面积RSD<4.8%。表明供试品溶液在3天内基本稳定。
5 指纹图谱建立
5.1 样品测定
按2.2项下方法分别制备11批供试品溶液,按2.3项下的电泳条件进样。样品图谱见图1。
5.2 共有指纹峰的确认及归属
通过比较生脉散与对照品的毛细管电泳图谱中峰的在线紫外吸收光谱和迁移时间,确认了五味子醇甲和五味子乙素。以五味子醇甲为参照物,计算11批生脉散电泳图中各峰的相对迁移时间,通过对相对迁移时间与成分的紫外光谱比较,确定了20个分离较好或峰面积较大的主要共有峰(图2)。各峰相对迁移时间、相对峰面积结果见表2、3。

图1 11批生脉散CE指纹图谱
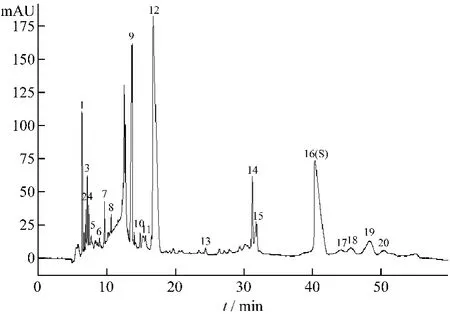
图2 生脉散CE指纹图谱(20个主要共有峰,峰16-五味子醇甲,峰19-五味子乙素)
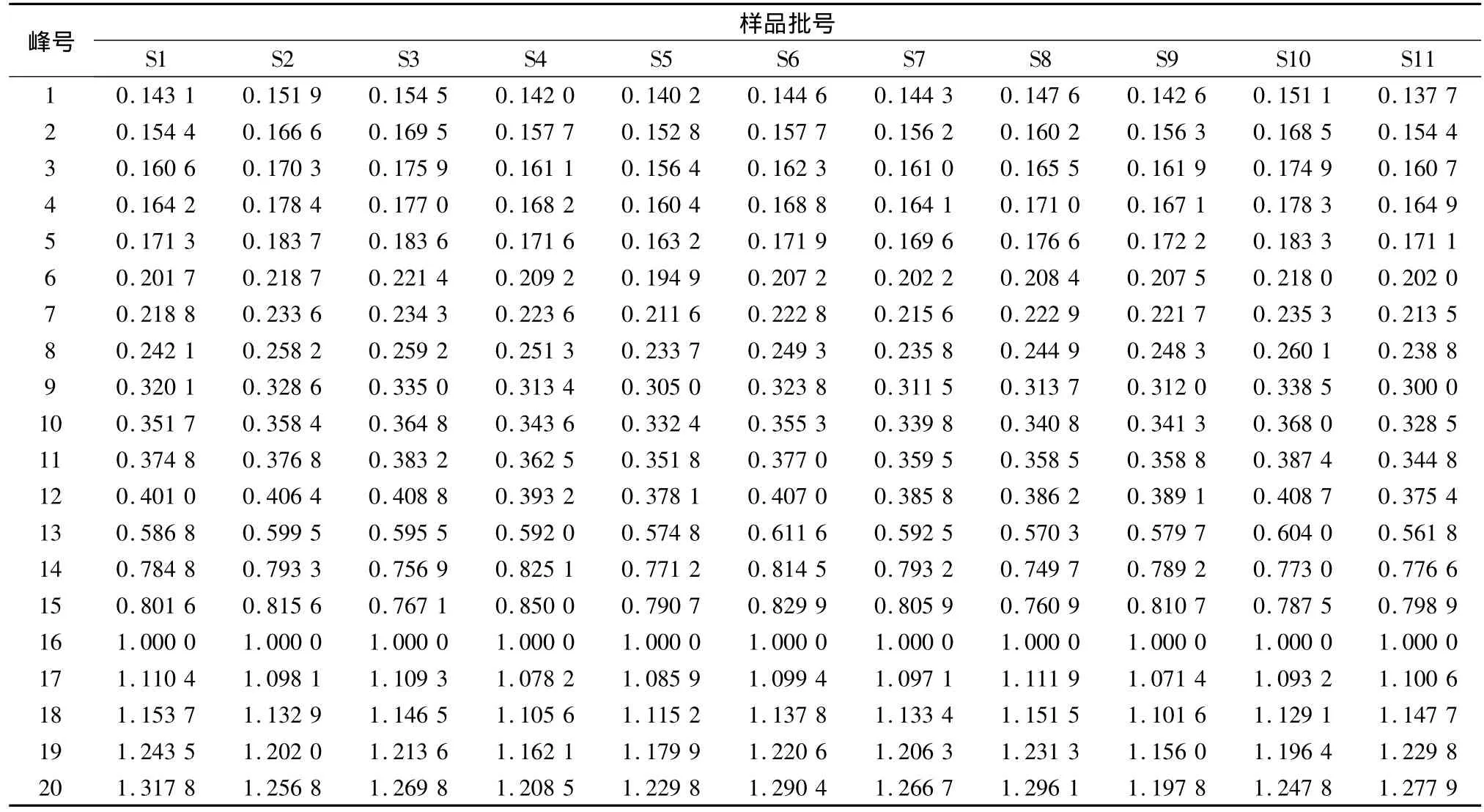
表2 生脉散共有峰的相对迁移时间
按2.3项下的实验条件建立单味药材和各药材阴性对照液的指纹图谱。根据峰的在线紫外光谱和迁移时间,对11批生脉散指纹图谱中主要共有峰的来源进行归属。20个主要共有峰中有4个峰来自红参(峰1、3、4、9),有6个峰来自麦冬(峰1、2、4、6、9、11),有 13 个峰来自五味子(峰 2、7、8、10、12、13、14、15、16、17、18、19、20),5 号峰为复方煎煮过程中新产生的成分。其中3个峰为红参和麦冬共有,1个峰为麦冬和五味子共有。
5.3 指纹图谱相似度评价
将所得的11批指纹图谱导入中药色谱指纹图谱相似度计算软件,进行峰匹配并生成生脉散指纹图谱的共有模式。以共有模式为标准,采用不同算法对每批样品进行相似度评价,结果见表4。在生成共有模式方法相同的条件下,夹角余弦法所得的相似度结果均略高于相关系数法的结果。相关系数方法主要反映两矢量间的变化趋势并可尽量减少背景截距的影响;而夹角余弦法则主要反映两矢量间的夹角余弦值,表现了两矢量间的形状差异。两种方法不存在本质差异,都是矢量的内积运算。建立的11批生脉散指纹图谱的相似度较高,表明药材成分较稳定,而且所采用的提取方法和优化的电泳条件具有良好的稳健性和可靠性。
6 讨论
6.1 药材提取方法的选择
比较了水提和70%乙醇提两种提取方法,结果发现水提取的样品溶液峰数多。临床上生脉散以煎煮成汤药的形式口服给药治疗疾病,为了更好地体现生脉散的药效物质成分,并保证与临床应用的一致性,本文选用水提取方法。
提取液经浓缩、稀释、离心、过滤后直接进样分析,不必经过有机溶剂的处理等操作,能最大限度地保留生脉散中的活性成分。
6.2 毛细电泳方法及其模式的选择
生脉散溶液采用反相高效液相色谱法分析时,即使梯度洗脱,分析时间长约120 min,而且许多组分在色谱柱中无保留、难以分离。此外,提取液中含有色素、蛋白质、糖类、鞣质等干扰物,色谱柱极易被污染从而使寿命变短。CE法能克服以上缺点,得到理想的分离效果,并且成本低廉。毛细管电泳的缓冲体系为水溶液,不受有机溶剂的背景干扰,检测波长可低至190 nm。
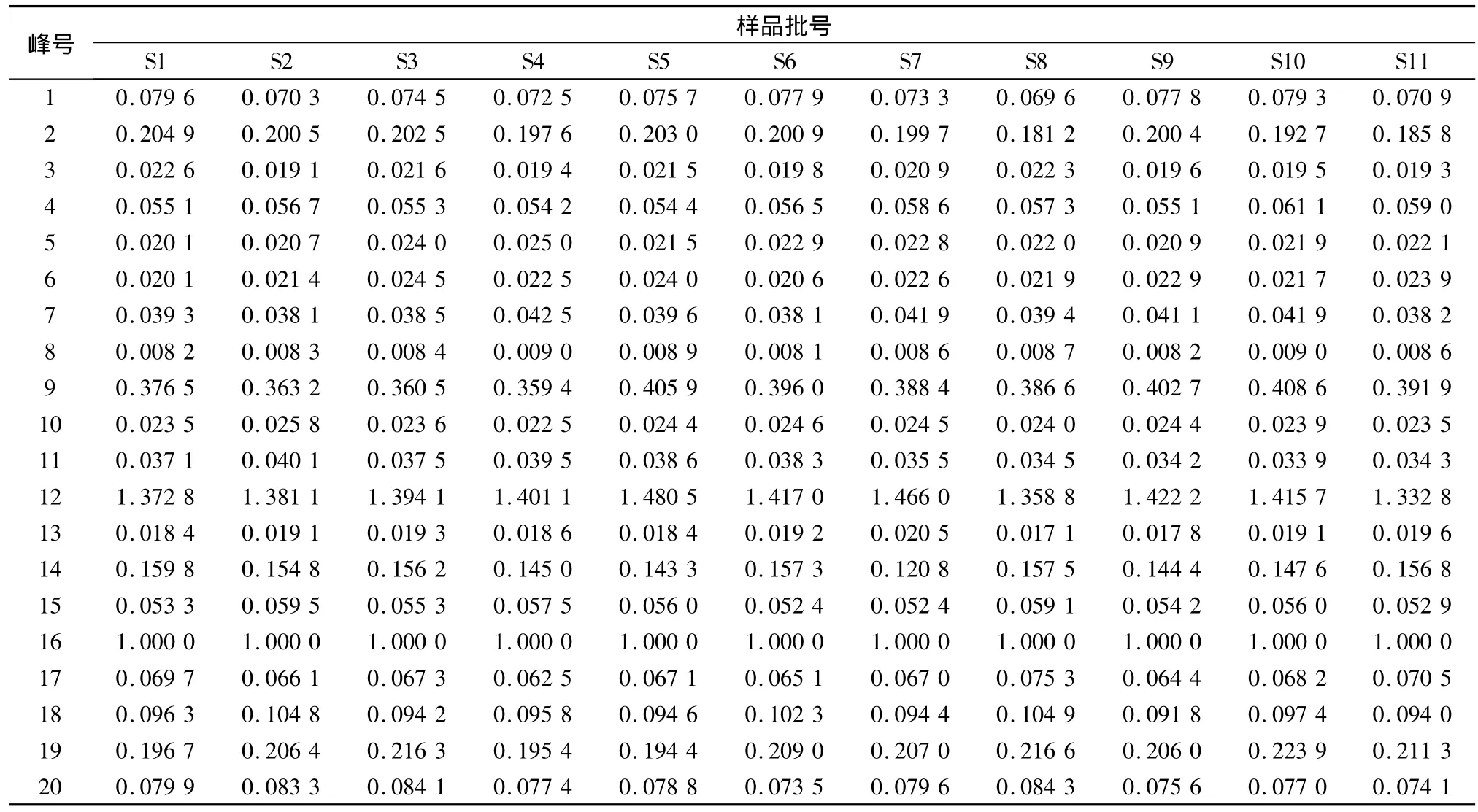
表3 生脉散共有峰的相对峰面积
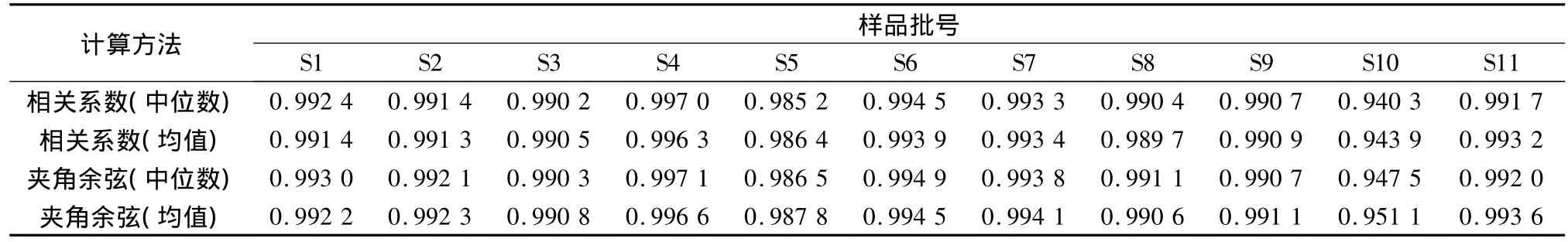
表4 不同算法的相似度计算结果
由于生脉散中有大量不带电荷的中性分子,在CZE模式下分离效果差,于是选用MEKC模式进行分离。硼砂电解质可与含有多羟基和邻位羟基的成分发生络合作用,使不同组分的淌度差异拉大,分离度增加,提高了CE的选择性。加入SDS可明显改善后面出峰组分的分离度。
CE分析时影响因素很多,同时不同因素对分离的影响程度存在明显的差异,而且生脉散成分众多、极其复杂,可见建立科学可靠的指纹图谱难度很大、工作量非常繁重。本文运用实验设计方法对一系列的实验因素同时进行考察,确定显著地影响分离的主要因素并优化其实验水平,获得最佳的分离条件。以尽量少的实验次数达到最优的实验结果,缩短方法建立的周期,达到事半功倍的效果。
本文方法稳定、可靠、重复性好。在建立生脉散指纹图谱的基础上,可结合药效学实验,进而研究指纹图谱与药效之间的相关性,更能为生脉散质量评价和控制提供依据。同时可构建研究复方多组分在体内吸收的血清指纹图谱,阐明其入血成分、代谢产物及方剂配伍意义,从而揭示生脉散的药效物质基础。
[1]谷凌云,王发渭,杨明会.生脉散类方治疗心血管疾病研究近况[J].军医进修学院学报,1997,18(2):150-152.
[2]Yao H T,Chang Y W,Chen C T,et al.Shengmai San reduces hepatic lipids and lipid peroxidation in rats fed on a high-cholesterol diet[J].J Ethnopharmacol,2008,116:49-57.
[3]You J S,Huang Huifeng,Chang Yingling,et al.Sheng-Mai-San reduces adriamycin-induced cardiomyopathy in rats[J].Am J Chin Med,2006,34(2):295-305.
[4]江 巍,阮新民,林 宇,等.生脉散对冠心病冠脉搭桥术后患者生存质量改善作用的临床观察[J].上海中医药杂志,2005,39(9):3-6.
[5]张群智.古方生脉散现代应用探讨[J].中成药,2001,23(4):286-288.
[6]朱丹妮,严永清,李志明.生脉散复方化学动态变化与药效关系的研究-生脉散复方化学的研究(Ⅲ)[J].中国中药杂志,1998,23(8):483-485.
[7]岳 磊,朱丹妮,严永清,等.生脉散复方化学动态变化与药效关系研究-生脉散中五味子化学动态变化(Ⅶ)[J].中国中药杂志,2006,31(12):1010-1012.
[8]Chang Y W,Yao H T,Chien D,et al.High-performance liquid
chromatography-electrospray mass spectrometry for the simultaneous determination of multiple components in Sheng-Mai San,a prescription of traditional Chinese medicine [J].Phytochem Anal,2008,19:258-265.
CE fingerprint of the compound Shengmai Powder
JI Yi-bing1, FAN Xiao-mei1,2, ZHU Dan-ni1
(1.China Pharmaceutical University,Nanjing 210009,China;2.The South China Center for Innovative Pharmaceuticals,Guangzhou 510663,China)
Shengmai Powder;capillary electrophoresis;fingerprint
AIM:To investigate the CE fingerprint of the compound Shengmai Powder(Radix et Rhizoma Ginseng rubra,Radix Ophiopogonis,Fructus Schisandrae chinensis).METHODS:Sequential uniform design was used to optimize the separation conditions.A CE fingerprint for Shengmai Powder was established using buffer comprising 44 mmol/L borate and 34 mmol/L SDS at pH 9.5,a running voltage of 25 kV,a capillary temperature of 25 °C and a wavelength of 200 nm.The sample was injected at a pressure of 50 mbar for 100 s.RESULTS:From the fingerprints of eleven batches of sample solutions,twenty main common peaks were determined.four peaks came from Radix et Rhizoma Ginseng,six peaks from Radix Ophiopogonis,thirteen peaks from Fructus Schisandrae chinensis,three peaks shared by Radix et Rhizoma Ginseng and Radix Ophiopogonis,one peak shared by Radix Ophiopogonis and Fructus Schisandrae chinensis and one new constituent.CONCLUSION:The developed method is accurate and reliable,and the fingerprint analysis can be used for the quality assessment and control of compound Shengmai Powder.
2009-11-17
国家自然科学基金资助项目(30772792)
季一兵(1970-),女,副教授,理学博士,研究方向:中药质量控制及现代仪器分析。Tel:(025)86185150 E-mail:jiyibing@jlonline.com
猜你喜欢
上海涂料(2021年5期)2022-01-15
世界最新医学信息文摘(2021年12期)2021-06-09
装备制造技术(2019年12期)2019-12-25
中成药(2018年12期)2018-12-29
中成药(2017年3期)2017-05-17
中成药(2017年3期)2017-05-17
中国粮油学报(2016年1期)2016-02-06
杂草学报(2015年2期)2016-01-04
药学研究(2015年11期)2015-12-19
中药与临床(2015年5期)2015-12-17
