
紫槐烧伤乳膏剂的制备及质量控制
2010-04-09 14:14:24熊维政姜家书
中国药业 2010年22期
熊维政,李 磊,石 磊,姜家书,左 杰
(河南羚锐制药股份有限公司,河南 新县 465550)
紫槐烧伤乳膏剂由紫草、虎杖、槐枝等组方,系民间老中医验方,具有清热凉血、收敛生肌、止痛功效,多年临床使用证明其对Ⅰ度和Ⅱ度烧烫伤具有良好效果。原处方以豚脂榨取药材、提取有效成分后制成油膏使用,制剂水平较低且使用不方便,制剂的质量控制比较困难。为更好地满足临床需要,笔者研制了紫槐烧伤乳膏剂,现将其制备工艺与质量控制方法报道如下。
1 仪器与试药
岛津LC-2010AHT高效液相色谱(HPLC)仪,SPD-20A紫外检测器(日本岛津公司);79-1型磁力加热搅拌器(金坛市医疗仪器厂)。紫草对照药材(批号为200802)、左旋紫草素对照品(批号为110769-200405)、冰片对照品(批号为110743-200504)、大黄素对照品(批号为110756-200110)均由中国药品生物制品检定所提供;紫槐烧伤乳膏剂(河南羚锐制药股份有限公司,批号为090913);紫草、虎杖、(张仲景大药房);冰片(云南省思茅市林缘香料有限公司);槐枝采自新县;自制纯化水;所用试剂均为分析纯;辅料均符合药用标准。
2 方法与结果
2.1 处方与制备
处方:紫草,虎杖,槐枝,冰片,硬脂酸,单硬脂酸甘油酯,液体石蜡,甘油,三乙醇胺,平平加O、羟苯乙酯,纯化水。
制备[1]:取虎杖、黄柏,粉碎成细粉,混匀,照2005年版《中国药典(一部)》附录ⅠO(流浸膏剂与浸膏剂)项下方法,以90%乙醇为溶剂,浸渍36h后缓缓渗漉,收集渗漉液600mL,滤过,滤液减压浓缩至相对密度为1.00的清膏,备用;取硬脂酸、单硬脂酸甘油醋、液状石蜡加热至80℃,溶解,搅匀,待温度降至50℃左右时加入上述醇提浸膏,搅拌均匀,作为油相,备用;另取甘油、三乙醇胺、平平加O、羟苯乙酯加热至80℃,充分熔化,搅匀后缓缓加入油相,沿一个方向搅匀(约100 r/min),待冷至40℃,加入冰片(乙醇适量溶解),用水调节至1 000 g,搅匀,分装即得。规格为每支15 g。
2.2 一般质量控制
性状:本品为均匀细腻的紫红色乳膏。
pH:取本品10 g,80℃加热熔化,加纯化水50mL稀释,溶液pH应为6.0~7.0。
其他:应符合2005年版《中国药典(一部)》软膏剂[1]项下有关规定。
2.3 鉴别
紫草[2]:称取供试品3.0 g、紫草对照药材粉末2.0 g、不含紫草的阴性对照品3.0 g,分别加氯仿30 mL,超声处理30 min后置分液漏斗中,将分液漏斗在10℃以下静置2h,分取氯仿液,作为供试品溶液、紫草对照药材溶液及阴性对照品溶液。取左旋紫草素对照品,加乙醇制成每1mL含1.0mg的溶液,作为对照品溶液。照薄层色谱法[1]试验,吸取上述4种溶液各10μL,分别点于同一硅胶G薄层板上,先以饱和15min的二氯甲烷-甲苯(5∶11)为展开剂上行展开,展距约为8.5 cm,取出,晾干,然后再以饱和15min的甲苯-乙酸乙酯-甲酸(5∶1∶0.1)为展开剂上行展开,展距约为3.5 cm,取出,晾干。供试品溶液色谱中,在与对照药材溶液和对照品溶液色谱相应位置上显相同的红褐色斑点,再喷以10%氢氧化钾甲醇溶液,斑点颜色变为蓝色,阴性对照品溶液色谱中则无此斑点(图1 A)。
虎杖[3]:在2.4含量测定项高效液相下色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
槐枝[4]:取供试品5.0 g、不含槐枝的阴性对照品5.0 g,分别加甲醇20mL,超声处理10min,滤过,滤液挥干,残渣加甲醇1mL使溶解,作为供试品溶液、阴性对照品溶液。取芦丁对照品用甲醇制成每1mL含1mg的溶液,作为对照品溶液。照薄层色谱法[1]试验,吸取上述3种溶液各10μL,分别点于同一硅胶G薄层板上,以饱和15min乙酸乙酯-甲酸-水(8∶1∶1)为展开剂上行展开,展距约为10 cm,取出,晾干,喷以三氯化铝乙醇溶液,于105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同的浅绿色斑点,阴性对照品溶液色谱中则无此斑点(图1 B)。
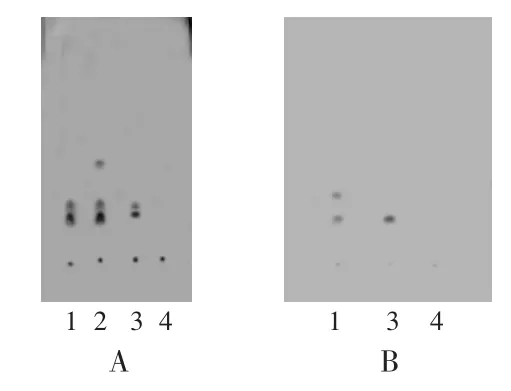
图1 薄层色谱图
冰片[3,5]:取本品2 g,加乙醇5mL搅拌溶解,加1%香草醛硫酸液2滴,即显紫色。
2.4 含量测定(虎杖)[3,6-7]
2.4.1 色谱条件与系统适用性试验
色谱柱:Agilent-C18柱(250mm×4.6mm,5μm);流动相:甲醇-0.1%磷酸溶液(80∶20);检测波长:254 nm;柱温:40℃;流速:1mL/min;进样量10μL。在该色谱条件下,对照品溶液及供试品溶液色谱峰分离良好,出峰时间合适(7.5min),阴性对照品溶液对测定无干扰(图2)。

图2 高效液相色谱图
2.4.2 溶液制备
称取供试品约3.0 g,精密称定,加入甲醇30mL及2.5mol/L硫酸溶液20 mL,加热回流60 min,用氯仿提取3次,每次10 mL,收集氯仿液,蒸干,残渣用甲醇溶解,转移至25mL量瓶中,甲醇定容至刻度,即得供试品溶液。另按处方制备不含虎杖的乳膏剂对照品,同法制备阴性对照品溶液。精密称取大黄素对照品6.4mg,置50 mL量瓶中,用甲醇充分溶解,定容至刻度,得贮备液备用,然后精密吸取贮备液1mL,置10mL量瓶中,用甲醇定容至刻度,得每1mL含大黄素12.8μg的溶液,作为对照品溶液。
2.4.3 方法学考察
线性关系考察:精密吸取对照品溶液2,4,6,8,10,12μL,注入色谱仪,以峰面积(A)对溶液质量浓度(C)进行线性回归,得回归方程 A=13 175X+129 604,R2=0.999 9(n=6)。结果表明,大黄素进样量在0.025 6~0.153 6μg范围内与峰面积线性关系良好。
精密度试验:精密吸取质量浓度为12.8μg/mL的大黄素对照品溶液10μL,注入液相色谱仪,重复进样5次。结果大黄素峰面积的 RSD为0.65%(n=5)。
稳定性试验:精密吸取供试品溶液10μL,分别于0,2,4,6,8,12,24h时进样测定。结果大黄素含量的 RSD为0.22%(n=7),表明供试品溶液在24h内稳定。
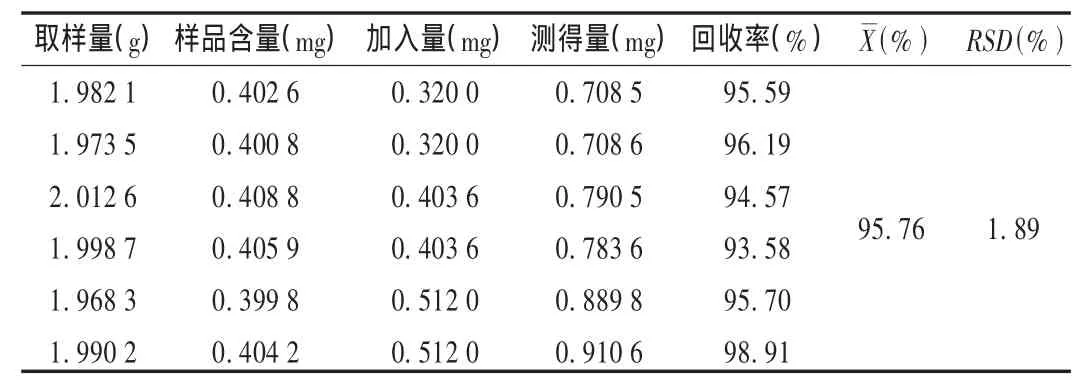
表1 大黄素加样回收试验结果(n=6)
加样回收试验:精密量取已知大黄素含量的样品6份(0.203 1 mg/g),加入一定量的大黄素对照品(按80%,100%,120%加入对照品,各加平行两份),按样品含量测定方法测定。结果见表1。
2.4.4 样品含量测定
取不同批号的样品3批,按供试品溶液制备方法制备溶液。分别精密吸取对照品溶液和供试品溶液各10μL,按上述色谱条件平行测定5次,按外标法以峰面积计算供试品中大黄素含量。结果批号为090913,090917,090919的样品中大黄素含量分别为0.201 2,0.211 7,0.193 4mg/g。因此,暂订每1 g本品含虎杖以大黄素计不得少于0.19mg/g。
2.5 稳定性考察
留样观察法:取本品3批,装入铝箔管内,密闭、避光、在恒温[(25±2)℃]、相对湿度(60±10)%的条件下储存6个月。结果样品外观、均匀度、稠度均无明显变化;每月测定的大黄素含量均符合规定,说明6个月内含量稳定。
离心法:取样品3批,各10 g,分别置离心管中,以3 000 r/min的转速离心处理。结果样品无分层现象。
3 讨论
本乳膏剂是在原油膏剂的基础上,通过科学的实验设计,采用乙醇渗漉法提得药物浸膏后,选择适宜的赋形剂制成的乳膏剂,与原来的油膏剂相比,药物有效成分的提取更完全,充分保证了临床疗效,制剂的外观性状也得到了改善,且使用方便,易于清洗,满足了临床患者的需要。
该制剂制备工艺简单,含量测定准确,制剂质量稳定,使用安全、方便,较油膏剂具有明显的优势。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:附录IO,附录IR,附录VIB,附录VID.
[2]张增巧,于 霖,吴延晖,等.复方紫归软膏的制备及质量控制[J].中国医院药学杂志,2000,20(1):47.
[3]洪求兵,梁晓霞,张孟佑,等.复方虎杖凝胶剂的制备及质量控制[J].中国药业,2008,17(15):54-55.
[4]王春桃,唐于平,周 玲.槐枝皮中黄酮类成分的研究[J].江苏中医药,2008,40(7):65-66.
[5]钟春元,黄英柱,方奕曦.复方大黄烧伤药膜的制备与应用[J].医药导报,2002,21(4):241.
[6]付艳敏,李伯军,白 岩.等.HPLC法测定肺结核丸中大黄素的含量[J].中国药师,2006,9(1):20-21.
[7]陈世虎,陈瑞龙,呼延玲.HPLC测定皮肤病血毒丸中大黄素与大黄酚含量[J].中国药师,2006,9(1):47-48.
猜你喜欢
中成药(2018年7期)2018-08-04 06:04:02
特别健康(2018年9期)2018-07-17 15:29:08
中成药(2018年3期)2018-05-07 13:34:47
中成药(2018年1期)2018-02-02 07:20:15
家教世界·创新阅读(2015年12期)2015-12-10 23:10:07
云南中医学院学报(2015年4期)2015-07-31 17:42:11
云南中医学院学报(2014年6期)2014-07-31 17:59:54
中国中医药现代远程教育(2014年15期)2014-03-01 04:27:48
中国中医药现代远程教育(2014年14期)2014-03-01 04:27:32
云南中医学院学报(2011年2期)2011-07-31 18:03:17
