
陕西省部分聋哑学生聋病易感基因分子流行病学研究△
2010-01-25 06:44朱一鸣郭玉芬刘晓雯王艳莉徐百成纪育斌历建强李倩王秋菊
听力学及言语疾病杂志 2010年3期
朱一鸣 郭玉芬 刘晓雯 王艳莉 徐百成 纪育斌 历建强 李倩 王秋菊
目前的研究认为,GJB2、SLC26A4基因和线粒体DNA12S rRNA m.1555A>G突变是导致非综合征型耳聋(non-syndromic sensorineural hearing loss, NSHL)的常见分子病因,并且在不同种族、不同地域的耳聋人群中有不同的突变热点和发病率[1],该三个基因已作为临床耳聋基因的常规检查。国内学者也在不同地区不同人群中进行了上述基因的流行病学研究[2~5]。有学者曾对陕西省部分聋哑学生进行了GJB2、SLC26A4基因及线粒体DNA12S rRNA m.1555A>G突变调查,三个基因的检测阳性率分别是19.23%(10/52)、19.64%(11/56,为c.919-2G>A杂合和纯合率)和1.72%(1/58)[3~5],但样本量偏小(分别为52、56、58例)。根据统计学抽样调查总体率样本量(N)估计公式N=4×P(1-P)/δ2,以上三个基因的发病率作为P值(估计值),δ(精度)分别设为5%、5%和2%,第一类错误概率α为0.05,进行了最小样本量预测,分别为249人(GJB2),252人(SLC26A4),169人(mtDNA12S rRNA m.1555A>G),在以上允许的误差范围内[6]。因此,本研究把样本量扩大到283人,对陕西省部分非综合征聋哑学生进行上述三个基因的联合筛查,以了解该地区聋哑人群常见致聋基因的热点突变及突变频率,为耳聋的防治提供分子病因依据。
1 资料与方法
1.1临床资料 选取陕西省四个特殊教育学校:汉中市特殊教育学校、咸阳市特殊教育学校、榆林市特殊教育学校以及神木县特殊教育学校的283名来自于不同家庭的非综合征型耳聋学生,汉族,其中男165人,女118人,男女比例为1.40:1;年龄2~27岁,平均12.23±4.79岁。
1.2方法
1.2.1病史采集 采用问卷调查及当场询问(家长或是老师)为主,电话家访为辅,内容主要包括:外耳和中耳疾病史、母孕期智力检查,有无异常和出生时是否缺氧、是否早产、耳聋前后有无明确头部外伤史、家族史、耳毒性药物的使用情况等,绘制相应的家系图谱,建立详尽的病历档案[7]。
1.2.2检查与采血 ①体检:全部对象均进行全身(智力、生长发育、是否合并其他系统疾病等)和专科检查(外耳内耳发育)以排除综合征型耳聋;②听力学检查:主要运用Madsen502便携式听力计进行纯音测听(PTA)或以美国智听公司Smart EP 听觉诱发电位仪检测声导抗和听性脑干反应(ABR)检查;③在知情同意的情况下抽取患者外周静脉血5~10 ml,ACD-EDTA抗凝,避免震荡、溶血,4℃运送至国家人类基因组北方研究中心保存。
1.2.3基因组DNA的制备与保存 用饱和氯化钠抽提法[8](盐析法)提取外周血白细胞DNA,TE溶解,1%琼脂糖电泳定性、紫外分光光度计定量和纯度检测,配制好的工作液4℃保存备用,原液-80℃长期保存。
1.2.4PCR扩增及酶切反应[2]GJB2基因和SLC26A4基因的第8和第19外显子的PCR产物进行双向直接测序,扩增767bp长度的mtDNA nt 1229~nt 1995目的片断,以Alw26Ι内切酶鉴定突变体,酶切阳性的样品进行直接测序。
1.2.5测序与生物学分析 所有测序均用双脱氧链终止法在ABI Prism 3700测序仪上直接测序, 将测序结果与标准序列进行比对, 确定基因突变位点。测序结果与均来自NCBI上的标准序列(GJB2: NG_008358.1;SLC26A4: NT_007933.15;mtDNA: NC_012920.1)在DNA Star 7.0和Sequencher 4.9上进行对比分析。
2 结果
2.1线粒体DNA12S rRNA m.1555A>G检测结果 283例患者共检测到9例m.1555A>G均质性突变,检出率为3.18%(9/283),所有携带m.1555A>G突变的患者同时发生了m.1438A>G位点的均质性多态改变,其中1例患者同时携带m.1555A>G均质性突变、m.1438A>G和m.1715C>T均质性多态改变。除9例均质性突变者外,还有1例患者同时携带m.1555A>G和m.1819C>T异质性碱基改变(图1)。
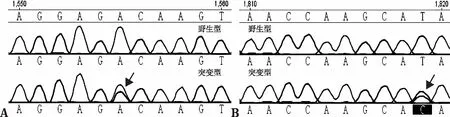
图1 mtDNA12S rRNA 两个异质性改变测序图A:m.1555A>G异质性突变,B:m.1819C>T异质性改变
2.2GJB2基因突变检测结果 283人中有55例患者明确为GJB2基因突变(包括纯合、复合杂合以及已知的显性突变)所致耳聋,占检测人数的19.43%(55/283)(表1);8例为突变携带者,另有133例检测到GJB2基因的其他碱基序列改变。表2为GJB2基因主要突变的等位基因发生频率和共检测到的17种碱基改变,包括11种致病突变(含2种显性致病:c.551G>A和c.224G>A)、4种多态、1种新突变(c.557C>T) 及1种存在争议的突变(c.109G>A)。其中,c.235delC和c.299_300delAT等位基因频率分别为14.13%(80/566)、3.89%(22/566),占所有致病等位基因数的87.18%(102/117),是该地区的热点突变。
2.3SLC26A4基因突变检测结果 通过外显子8和19的检测,7例(2.47%,7/283)患者为SLC26A4基因双等位基因突变(纯合和复合杂合突变),均为c.919-2A>G和c.2168A>G(p.H723R)突变;13例携带有SLC26A4基因单等位基因碱基改变,其中,有三种其他突变c.919-18T>G、c.920C>T(p.T307M)和c.1001+32A>G(表3)。c.919-2A>G和c.2168A>G(p.H723R)占所有等位基因数的88.89%(24/27)(表4)。

表1 GJB2基因致病突变患者统计(例)
注:*已知的显性突变(数据来源:http://davinci.crg.es/deafness/index.php)。本表不包括多态和可疑致病突变
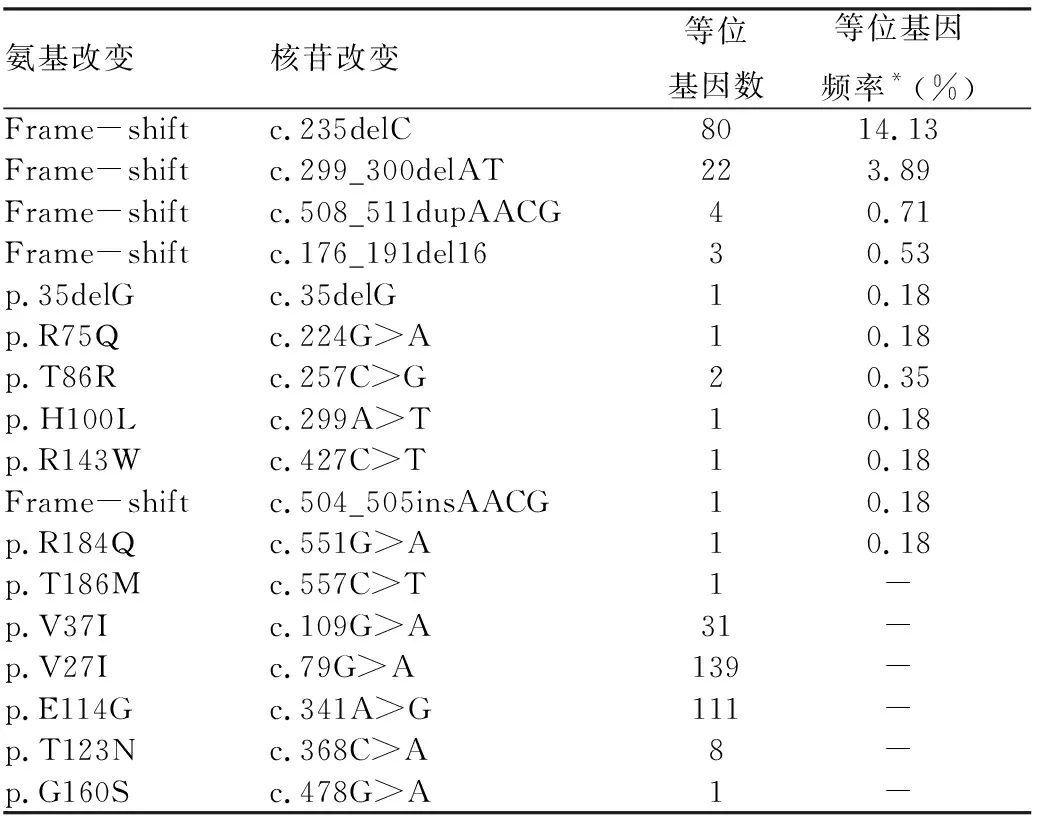
表2 GJB2基因等位基因突变频率统计
注:*占整个等位基因(283×2=566)的比例,不确定致病性和多态核苷改变不做统计

表3 SLC26A4基因碱基改变及人数(例)

表4 SLC26A4基因等位基因统计
3 讨论
由于人种和地域的不同,GJB2、SLC26A4基因及mtDNA12S rRNA m.1555A>G突变在不同人种、不同国家的耳聋人群中的发病频率及突变热点都不同[1]。在国内,由于样本量的问题,如何得到三个基因在不同地区不同人群中的确切发病率一直是个值得关注的问题[6],本研究在允许误差分别为5%、5%和2%的情况下,应用最小样本量的采样方法得到GJB2、SLC26A4基因及mtDNA12S rRNAm.1555A>G突变在陕西聋哑学生的发病率分别是19.43%(55/283)、2.47%(7/283)、3.18%(9/283),GJB2突变率与既往报道类似;mtDNA12S rRNA m.1555A>G的突变率高于既往的报道,SLC26A4的突变率明显低于既往国内文献的报道[既往报道单纯c.919-2A>G的纯合突变比率就达到10.7%(6/56)][4],由此可见样本量的偏差可导致突变比例明显不同。
本研究中,GJB2基因的阳性检测率与以往报道的该地区的阳性率基本接近[3],c.235delC及c.299_300delAT为陕西地区GJB2基因主要突变形式也与以往报道吻合,可见,GJB2基因突变导致的先天性耳聋是该地区聋哑人群最主要的组成部分,所以对GJB2基因携带者进行婚配指导是有效降低该地区耳聋人群的主要方法之一。Richard[9]和Hamelmann[10]曾对p.R75Q(c.224G>A)和p.R184Q(c.551G>A)突变致聋的常染色体显性遗传家系进行过详细报道。本研究也发现GJB2基因c.224G>A(p.R75Q)和c.551G>A(p.R184Q)突变分别与两个常染色体显性耳聋家系密切相关。
目前的研究显示,在中国人群中SLC26A4第8、第19外显子的c.919-2A>G和c. 2168A>G突变为热点突变,尤其是c.919-2A>G[11]。因此,本研究针对上述两个外显子进行分析,结果发现,双等位基因(纯合和复合杂合)突变频率为2.47%,与既往报道[4]不同,分析其原因,除样本量差别外,由于SLC26A4基因突变是大前庭水管综合征(large vestibul araqueduct syndrome,LVAS)的分子病因,LVAS是一种先天性内耳畸形疾病,但患儿出生时听力损失并不存在或尚未出现,多为出生后几年发病,多数患儿已经有了完整的语言体系,而且患者听力下降为波动性,因此,患儿会佩戴助听设施,很少在聋哑学校就读,因此,这也是SLC26A4基因在聋哑学校学生中检出率较低的可能原因。
有研究显示,线粒体DNA12S rRNA m.1555A>G突变在中国西北地区(甘肃和青海)有较高的发病率[2, 12],研究者认为由于上述地区经济文化落后,抗生素滥用及近亲结婚或少数民族局部区域内通婚是导致该突变频率较高的可能原因。本研究中,mtDNA12S rRNA m.1555A>G突变频率为3.18%(9/283),较甘肃、青海地区低,考虑可能原因为该地区主要为汉族,少数民族人口较少,局部区域或民族内通婚较少。本研究同时还发现1例患者同时携带m.1555A>G和m.1819C>T异质性改变,该患者的同胞姐姐亦为先天性聋患者,但mtDNA12S rRNA m.1555A>G检查正常;m.1819C>T位于线粒体16S rRNA,该核酸改变是否致病、单独致病或是联合m.1555A>G同时致病,目前尚未确定[13,14]。
本研究进一步强调了应该尽可能按照流行病学的方法合理选择样本量,同时,可以明确陕西地区部分特教学校聋生25.08%(GJB2:19.43%,SLC26A4:2.47%, m.1555A>G:3.18%)的耳聋病因,有利于对该地区聋哑患者及其家系成员进行有效的预防、治疗及干预措施,并提供遗传学指导。
(致谢:感谢兰州易听医疗设备有限公司和四所特教学校所有教职员工以及学生家长在本课题研究中给与的大力支持和帮助。)
4 参考文献
1 Bayazit YA, Yilmaz M. An overview of hereditary hearing loss[J]. ORL J Otorhinolaryngol Relat Spec, 2006, 68: 57.
2 Guo YF, Liu XW, Guan J, et al. GJB2, SLC26A4 and mitochondrial DNA A1555G mutations in prelingual deafness in Northern Chinese subjects[J]. Acta Otolaryngologica, 2008, 128: 297.
3 Dai P, Yu F, Han B, et al. GJB2 mutation spectrum in 2063 Chinese patients with nonsyndromic hearing impairment[J]. Journal of Translational Medicine, 2009, 7: 26.
4 Dai P, Li Q, Huang DL, et al. SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss[J]. Genetics in Medicine, 2008, 10: 586.
5 朱庆文, 刘新, 韩东一, 等. 西北五省573例非综合征型耳聋患者线粒体DNA A1555G突变分析[J]. 临床耳鼻咽喉头颈外科杂志, 2007, 21: 460.
6 纪育斌, 韩东一, 王大勇, 等. 山东省聋哑学校485例耳聋患者易感基因突变检测分析[J]. 中华医学杂志, 2009, 86:2 531.
7 王秋菊, 杨伟炎, 韩东一.遗传性耳聋资源的收集、保存及利用-综合系统的建立[J]. 中华耳科学杂志, 2003, 1: 65.
8 Miller SA, Dykes DD,Polesky HF.A simple salting out procedure for extracting DNA from human nucleated cells[J]. Nucleic Acids Research, 1988, 16: 1 215.
9 Richard G, White TW, Smith LE, et al. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma[J]. Human Genetics, 1998, 103: 393.
10 Hamelmann C, Amedofu GK, Albrecht K, et al. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana[J]. Human Mutation, 2001, 18: 84.
11 Wang QJ, Zhao YL, Rao SQ, et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China[J]. Clinical Genetics, 2007, 72: 245.
12 惠培林, 郭玉芬, 鲍晓林, 等. 青海省部分聋校学生线粒体DNA12SrRNAA1555G和GJB2基因突变筛查报告[J]. 听力学及言语疾病杂志, 2009, 17: 23.
13 Khrapko K. Two ways to make an mtDNA bottleneck[J]. Nature Genetics, 2008, 40: 134.
14 Chinnery Pf, Howell N, Lightowlers RN, et al. MELAS and MERRF. The relationship between maternal mutation load and the frequency of clinically affected offspring[J].Brain, 1998, 121: 1 889.
猜你喜欢
内蒙古统计(2021年4期)2021-12-06
中医眼耳鼻喉杂志(2021年1期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
活力(2019年11期)2019-07-20
中国卫生统计(2019年3期)2019-07-10
快乐语文(2018年35期)2018-11-29
中国生殖健康(2018年4期)2018-11-06
华人时刊(2016年19期)2016-04-05
中国中医药现代远程教育(2014年13期)2014-03-01
中国卫生统计(2012年1期)2012-12-04
