
散发型亨廷顿病的临床表型及基因分析
2010-01-16 09:25王育新张本恕
天津医药 2010年11期
王育新 张本恕
散发型亨廷顿病的临床表型及基因分析
王育新 张本恕
亨廷顿病 基因 诊断 聚合酶链反应 表型 棘红细胞
亨廷顿病(Huntington disease,HD)是 IT15 基因 5′末端CAG重复序列不稳定扩展所致。HD致病基因的发现有助于对缺乏家族史的散发型HD患者做出正确诊断。本研究对11例临床诊断的散发型HD患者进行基因学分析,报告如下。
1 资料与方法
1.1一般资料 11例散发型HD患者系2003年6月—2008年12月我院和天津市环湖医院神经内科锥体外系门诊患者,均符合以下临床三联症(舞蹈、痴呆和人格障碍),无阳性家族史。其中男8例,女3例。平均发病年龄(48.3±7.7)岁。每例研究对象采静脉血2 mL,乙二胺四乙酸(EDTA)抗凝用于基因检测。
1.2临床表型的评定 采用HD统一评定量表(unified Huntington disease rating scale,UHDRS)[1]对患者的运动、精神行为和独立性3个方面进行评分,采用简易智能精神状态量表(mini-mental state exam,MMSE)评定患者认知功能。外周血涂片检查棘红细胞。
1.3IT15基因CAG重复次数的检测
1.3.1基因组DNA提取 按照DNA提取试剂盒(北京赛百盛基因技术有限公司)说明书进行。
1.3.2目的基因的体外扩增 上游引物5′-CCCAGAGCCCC ATTCATTGCC-3′,下游引物 5′-GGCGGCGGTGGCGGCTGT TGC-3′。PCR 反应体系:共 25 μL,其中 10×PCR Buffer 2.5 μL,2.5 mmol/L dNTP 2 μL,上、下游引物(10 μmol/L)各 1 μL,二甲基亚砜 2.5 μL,模板 DNA 0.5~1 μg,Taq DNA 聚合酶1 U。扩增条件:94℃预变性5 min,94℃变性60 s,64℃退火60 s,72℃延伸60 s,共30个循环,最后72℃延伸5 min。于2%琼脂糖凝胶电泳检测扩增结果。
1.3.3CAG重复次数的检测 PCR扩增产物经8%非变性聚丙烯酰胺凝胶电泳和硝酸银染色,参照DNA marker,估计PCR扩增产物的长度。参照基因序列图,CAG重复次数=(PCR产物长度-164)/3。
2 结果
11例散发型HD患者中5例具有扩展的CAG重复序列,见图1。5例确诊的散发型HD患者的发病年龄38~62岁,平均(47.1±9.2)岁;致病的 CAG 重复次数为 40~48次,平均(44.0±3.2)次;运动评分平均(33.6±8.3)分;独立性评分平均(86.0±8.9)分。MMSE平均(27.0±1.0)分。3例有精神和行为改变,表现为情绪低落、易激动或强迫行为。外周血涂片检查未见棘红细胞。
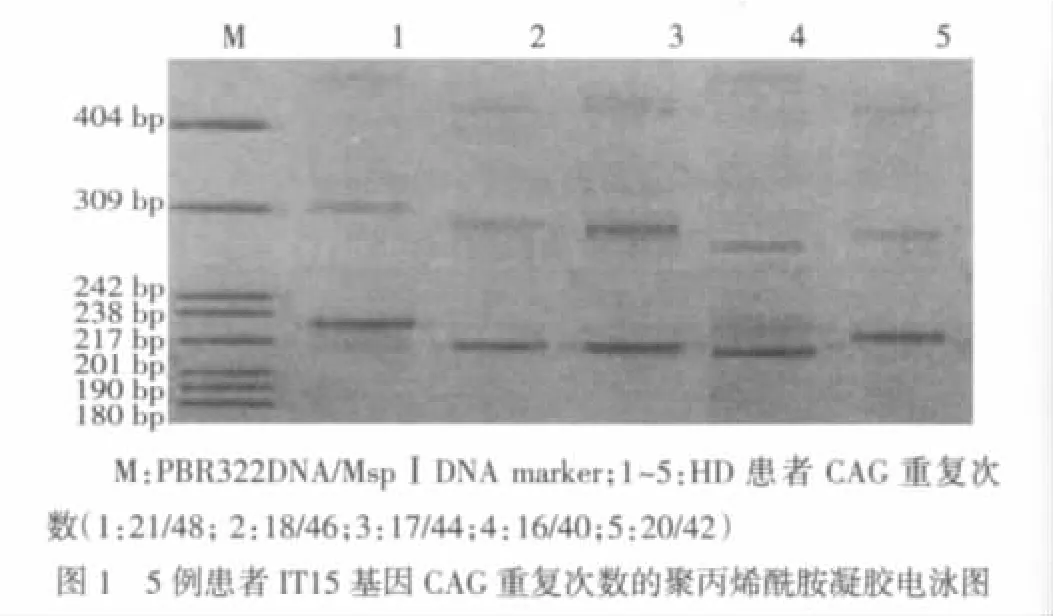
3 讨论
本研究经基因诊断确诊的5例散发型HD患者发病年龄偏大,平均47岁,CAG重复40~48次,轻度舞蹈,无明显智能障碍。中国人散发型HD的表现与国外的研究基本一致[2]。有家族史的HD患者发病年龄偏小,平均42岁,CAG重复 40~62 次,中~重度舞蹈,轻~中度智能障碍[3]。Durr等[4]发现2例散发型患者父亲的CAG重复向子代传递的过程中,由41次扩展为51次。无家族史的原因是由于疾病的早现。Davis等[5]检测1例散发型HD患者的父亲CAG重复35次。CAG重复位于27~35次的中间等位基因的不稳定扩展是造成新突变的主要原因。家族史缺乏的其他原因包括父母的早亡、误诊或被收养等。本文5例确诊的HD患者不存在被收养,3例父母因冠心病和脑血管病已去世,另2例患者的父母不同意基因检测。中国人散发型HD的病因尚不能除外疾病的早现或中间等位基因的不稳定扩展。
HD患者精子的CAG重复次数与外周血白细胞不同,存在体细胞嵌合性[6]。对于HD患者,受累的组织不易获得,外周血白细胞是获取DNA的唯一来源,因此无CAG重复的散发型HD患者不能排除体细胞嵌合的可能性。有研究发现多种Huntingtin相关蛋白结合到Huntingtin的多聚谷氨酰胺链,影响到其功能,进一步改变了细胞功能[7]。这些无CAG重复患者的HD基因内可能有尚未发现的其他类型的突变,或者由于编码Huntingtin相关蛋白的基因发生变化,目前尚未发现这种分子水平上的遗传异质性的证据。
Huntington disease-like 2 (HDL-2) 是 junctophilin3(JPH3)基因的CTG/CAG重复扩展所致,具有与HD相似的临床表型。Rosenblatt等[8]报道8例患者的临床表型与HD相似,无IT15基因的CAG重复扩展,也未发现与DRPLA、MJD和SCA-1、2、6相关的三核苷酸重复扩展。这些患者称为HD拟表型,或许由未知的三核苷酸重复扩展引起。笔者将继续对6例具有与HD相似的临床表型而无CAG重复扩展的患者进行以上疾病的三核苷酸重复扩展的检测,以期对这些患者做出进一步的诊断。
[1] Huntington Study Group.Unified Huntington’s disease rating scale reliability and consistency[J].Movement Disorders,1996,11(2):136-142.
[2] 王育新,张本恕.IT15基因CAG重复次数与亨廷顿病临床表型的相关性研究[J].中华神经科杂志,2007,40(7):593-602.
[3] Nance MA,Westphal B,Nuget S,et al.Diagnosis of patients presenting to a Huntington disease(HD)clinic without a family history of HD[J].Neurology,1996,47(6):1578-1580.
[4] Durr A,Dode C,Hahn V,et al.Diagnosis of sporadic Huntington’s disease[J].J Neurol Sci,1995,129(1):51-55.
[5] Davis MB,Bateman D,Quinn NP,et al.Mutation analysls in patients with possible but apparently sporadic Huntington’s disease[J].Lancet,1994,344(8924):714-717.
[6] Wheeler VC,Persichetti F,McNeil SM,et al.Factors associated with HD CAG repeat instability in Huntington disease[J].Med Genet,2007,44(11):695-701.
[7] Li XJ,Friedman M,Li S.Interacting proteins as genetic modifiers of Huntington disease[J].Trends Genet,2007,3(11):531-533.
[8] Rosenblatt A,Ranen NG,Rubinsztein DC,et al.Patients with features similar to Huntington’s disease without CAG expansion in huntingtin[J].Neurology,1998,51(1):215-220.
300052 天津医科大学总医院神经内科(2010-03-10收稿 2010-05-10修回)(本文编辑 闫娟)
猜你喜欢
肝博士(2022年3期)2022-06-30
中国听力语言康复科学杂志(2021年6期)2021-12-21
新生代(2019年15期)2019-10-21
科学24小时(2019年4期)2019-06-10
文体用品与科技(2019年9期)2019-05-08
电影(2019年3期)2019-04-04
医药前沿(2019年29期)2019-01-05
风湿病与关节炎(2018年1期)2018-02-11
科学生活(2016年7期)2016-07-25
心血管病学进展(2015年6期)2015-02-22
