
N-乙酰神经氨酸中通过抑制Nrf2轴促进缺氧/复氧损伤的H9C2心肌细胞发生铁死亡
2025-02-06 00:00:00季春斐左宗超王钧李妙男
南方医科大学学报 2025年1期
摘要:目的 基于铁死亡探讨N-乙酰神经氨酸(Neu5Ac)加剧H9C2大鼠心肌细胞缺氧/复氧损伤(H/RI)的影响及作用机制。方法 以大鼠心肌细胞株H9C2细胞作为研究对象,将细胞分为Control组、H/R 0 h组、H/R 3 h组、H/R 6 h组、H/R 9 h组、H/R 12 h组和H/R 15 h组,在缺氧缺糖8 h,分别复氧复糖0、3、6、9、12、15 h建立细胞缺氧/复氧损伤模型模拟心肌缺血再灌注损伤,通过实验选择最佳复氧时间进行后续实验。根据最佳复氧时间,复氧时给予不同Neu5Ac浓度的完全培养基作为复氧液,将细胞分为Control、0、5、10、20、30、40、50、60 mmol/L组,通过实验选择最佳药物浓度进行后续实验。根据前述最佳复氧时间和最佳给药浓度探讨Neu5Ac对H9C2心肌细胞加剧损伤的机制,将H9C2心肌细胞分为5组:Control组、H/R组、H/R+Neu5Ac组、H/R+Fer-1(铁死亡抑制剂)组、H/R+Neu5Ac+Fer-1组。通过检测5组细胞超氧化物歧化酶(SOD)活性,判断各组细胞氧化应激水平;使用FerroOrange 荧光探针和C11 BODIPY 581/591 荧光探针分别检测细胞内Fe2+和脂质过氧化物(Lipid ROS)水平;此外,通过Western blotting 检测Neu5Ac 在H9C2 心肌细胞H/RI 中对核因子E2 相关因子2(Nrf2)、谷胱甘肽过氧化物酶4(GPX4)、血红素加氧酶-1(HO-1)、铁死亡抑制蛋白1(FSP1)、胱氨酸/谷氨酸反向转运体(xCT)蛋白表达的影响。结果 H9C2 心肌细胞缺氧缺糖8 h 后,H/R 6 h 组和除H/R 9 h 组之外的其他组相比,Nrf2、GPX4、HO-1、FSP1 蛋白表达最少,且差异有统计学意义(Plt;0.001)。与Control组相比,不同浓度药物的实验组细胞活力均明显降低(Plt;0.0001),且随药物浓度逐渐增大,细胞活力逐渐减小,并测得该药物的半抑制浓度IC50=30.07 mmol/L。选择心肌细胞未发生明显铁死亡的时间,即缺氧缺糖8 h合并复氧复糖3 h,且复氧复糖时Neu5Ac浓度为30 mmol/L进行后续实验。结果显示,Neu5Ac可以提高SOD活性,增加Fe2+和Lipid ROS水平,降低Nrf2、GPX4、xCT、HO-1、FSP1等5种蛋白的表达(Plt;0.05)。在Neu5Ac的基础上使用Fer-1后,与H/R+Neu5Ac组相比,SOD 活性下降,Fe2+和Lipid ROS 水平减少,Nrf2、GPX4、xCT、HO-1、FSP1 等5 种蛋白的表达升高(Plt;0.0001)。结论Neu5Ac在H/RI中加剧心肌细胞发生铁死亡可能是通过抑制Nrf2轴进而产生过多的活性氧和脂质活性氧而导致。
关键词:N-乙酰神经氨酸;心肌细胞;缺氧/复氧损伤;铁死亡
国家心血管病中心于2024 年7 月发布的《中国心血管健康与疾病报告2023 概要》显示,在我国城乡居民疾病死亡构成比中,心血管病占首位,且我国每5 例因病死亡病例中就有2 例死于心血管病[1]。近年来,随着社会的发展和老龄化的加剧,缺血性心脏病成为人类致死致残的重要原因[2]。临床研究发现,约有15%的患者在成功血运重建后出现胸痛、心衰、再发心肌梗死、支架内血栓、支架内再狭窄等主要不良心血管事件(MACEs)[3]。即使临床经历了血运重建治疗,缺血心肌恢复血液再灌注后,患者病情仍有可能出现恶化,引起心肌超微结构、功能、代谢及生理结构发生进一步损伤,这种现象称为心肌缺血-再灌注损伤(MIRI)[4]。因此,作为体内不可再生细胞之一的心肌细胞,如何减轻MIRI仍然是临床治疗中面临的难题。
2019 年,有研究在国际上首次揭示铁死亡是心脏损伤的重要靶点和分子机制,并且在以小鼠为实验对象建立在体心肌缺血再灌注模型上,进一步证实给予铁死亡抑制剂或铁螯合剂可明显减轻缺血再灌注导致的急性和慢性心脏损伤[5]。既往研究表明,铁死亡抑制剂、铁螯合剂、线粒体特异性抗氧化剂等可以缓解心肌细胞铁死亡[6, 7],而有研究显示,神经氨酸酶1(NEU1)抑制剂,如扎那米韦(瑞乐砂)和奥司他韦(达菲),不仅用于流感的治疗,还是限制缺血再灌注损伤后心肌损伤和功能障碍的新疗法[8]。因此,神经氨酸酶1 与铁死亡和MIRI 存在怎样的关系,值得探索。
前期研究结果显示,在疾病状态下,神经氨酸酶(NEUs),又称为唾液酸酶,可以与弹性结合蛋白(EBP)、保护蛋白/组织蛋白酶A(PPCA)形成弹性蛋白受体复合物(ERC),发挥催化作用发生去唾液酸化,将糖蛋白和糖脂的末端糖——N-乙酰神经氨酸(Neu5Ac)游离下来,游离的Neu5Ac不仅可以导致细胞间的异常黏附以及跨膜信号传导的紊乱[9, 10],还可以通过促进炎症反应、氧化应激等多种途径损伤冠脉血管内皮,促进冠脉粥样硬化,作为神经氨酸酶的下游代谢产物[11, 12],目前已经证实,血浆 Neu5Ac水平是冠心病进展过程中高度升高的一个代谢物[13-16]。此外,本课题组前期研究结果也显示, Neu5Ac的水平对急性冠脉综合征(ACS)的诊断、危险分层以及ACS患者的临床预后具有重要的参考价值[17, 18]。
MIRI时,心肌细胞代谢出现异常,其机制涉及氧化应激、炎症反应、线粒体能量代谢障碍等[19, 20]。目前为止,尽管有研究表明NEU1 可以促进MIRI 损伤,但对NEU1的下游产物——Neu5Ac在 MIRI中对心肌细胞的直接损伤作用及其机制鲜有探索。在此背景下,本研究以H9C2心肌细胞为对象,在体外建立缺氧/复氧损伤(H/RI)模型,探讨Neu5Ac在H/RI中潜在途径及其分子机制,阐述Neu5Ac水平与ACS不良预后相关性的可能内在机理,为拓宽临床MIRI干预药物——神经氨酸酶抑制剂在MIRI防治的应用中提供新的理论依据和新的切入点。
1 材料和方法
1.1 实验细胞
H9C2大鼠胚胎心肌细胞株(普诺赛)。
1.2 主要试剂
N-乙酰神经氨酸、Ferrostatin-1(MCE);DMEM高糖基础培养基(gibco);DMEM 无糖基础培养基(Servicebio );胰蛋白酶、胎牛血清(普诺赛);青霉素-链霉素溶液(biosharp);Nrf2抗体、GPX4抗体、xCT抗体、HO-1 抗体、β‑actin 抗体(Affinity);FSP-1 抗体(proteintech);Goat Anti-Rabbit IgG(H+L)HRP 抗体(Affinity)CCK-8 试剂盒(Beyotime);超氧化物歧化酶(SOD)测定试剂盒(建成生物);C11 BODIPY 581/591(ABclonal);FerroOrange荧光探针(DOJINDO)。
1.3 主要仪器
全波长酶标仪(Thermo Fisher),梯度凝胶生成系统(北京君意华鑫公司);二氧化碳培养箱、三气培养箱(Thermo Fisher);超高分辨率激光共聚焦显微镜(Olympus)。
1.4 H/RI模型建立及分组
取在二氧化碳细胞培养箱常规培养(5%CO2,95%空气,37 ℃)、处于对数生长期且密度为80%左右的H9C2心肌细胞进行实验。根据培养条件及复氧不同的时间将细胞分为7组:Control组、H/R 0 h组、H/R 3 h组、H/R 6 h 组、H/R 9 h 组、H/R 12 h 组、H/R 15 h 组。去除旧培养基,用PBS润洗3遍,实验组用无糖无血清培养基替代原完全培养基,置于三气培养箱(94%N2,5%CO2,1%O2,37 ℃)中培养8 h,缺氧培养结束后,用完全培养基置换缺氧液,于二氧化碳细胞培养箱(5%CO2,95%空气,37 ℃)分别培养0、3、6、9、12、15 h。其次,将细胞均匀种于两个96孔板中,每组6个复孔。根据复氧时给予不同药物浓度的完全培养基作为复氧液,将细胞分为9 组:Control 组、0、5、10、20、30、40、50、60 mmol/L组。待H9C2细胞贴壁并且融合度约为80%时,将实验组的完全培养基置换为缺氧液,在三气培养箱进行缺氧实验。缺氧实验结束后,置换为100 μL含不同药物浓度的完全培养基在二氧化碳培养箱进行复氧实验。在复氧实验结束前2 h,弃旧复氧液,每孔置换为100 μL不同药物浓度的完全培养基+10 μL CCK-8溶液,于37 ℃二氧化碳培养箱继续培养2 h 后,全波长酶标仪检测吸光度A450 nm 值。将细胞随机分为5 组,Control 组、H/R组、H/R+Neu5Ac 组(H/R+N组)、H/R+Fer-1 组(H/R+F组)、H/R+Neu5Ac+Fer-1 组(H/R+N+F组)。实验组先用完全培养基在二氧化碳细胞培养箱常规培养,待细胞融合度达80%左右时,置换为无糖无血清基础培养基,在三气细胞培养箱缺氧培养8 h,复氧液分别为完全培养基、Neu5Ac(30 mmol/L)、Fer-1(10 μmol/L)、Neu5Ac(30 mmol/L)和Fer-1(10 μmol/L)的混合液。
1.5 CCK-8检测细胞活性
将H9C2细胞按照3000/孔种于96孔板中,按照上述分组进行H/RI模型及给药处理。复氧实验结束前2 h,弃旧复氧液,每孔置换为100 μL不同药物浓度的完全培养基+10 μL CCK-8 溶液,于37 ℃二氧化碳培养箱继续培养2 h后,酶标仪检测吸光度A450 nm值。
1.6 细胞SOD活性检测
将H9C2 细胞按照80×104/皿接种于直径10 cm培养皿中,按照上述分组进行H/RI 模型及给药处理。PBS 洗涤3 次,加入60 μL 高强度裂解液,冰上裂解15~20 min。用细胞刮刀刮下细胞,移入1.5 mL EP管中,4 ℃、12 000 r/min,20 min,进行离心。取上清液于新的1.5 mL EP 管中,BCA法测定各组细胞的总蛋白浓度后,将正常组样本稀释成不同浓度,按照说明书配制反应液,37 ℃培养箱孵育20 min 后,酶标仪检测吸光度A450 nm值。选取SOD抑制率在40%~60%的这一组稀释的正常组细胞,按照此比例稀释细胞H/R组、H/R+N组、H/R+F组、H/R +N+F组,进行正式批量试验。
1.7 细胞中Fe2+的检测
在无菌共聚焦小皿底部加入300 μL约含3×104细胞的混悬液,培养箱中放置2 h,待细胞沉降贴壁后在皿中加入1 mL完全培养基,待细胞汇合度达到80%左右时,进行缺氧/复氧处理。PBS洗涤3次,将500 μL浓度为2 μmol/L的FerroOrange工作液铺满小皿底部,37 ℃培养箱孵育30 min。无需清洗,利用活细胞成像系统收集相关荧光信号进行成像,并利用ImageJ 软件进行荧光强度分析。
1.8 细胞内Lipid ROS检测
在无菌共聚焦小皿中种皿以及缺氧/复氧处理步骤同1.7。PBS 洗涤3 次,将500 μL浓度为10 μmol/L 的C11 BODIPY 581/591 荧光探针,铺满小皿底部,37 ℃培养箱孵育1 h。PBS洗涤细胞3次,设置超高分辨率激光共聚焦显微镜的激光器激发波长为488 nm和561 nm,收集相关荧光信号进行成像,并利用ImageJ 软件进行荧光强度分析。
1.9 Western blotting检测
将H9C2细胞按照80×104 /皿种于直径10 cm培养皿中,按照上述分组进行H/RI模型及给药处理。提取细胞总蛋白,BCA法检测蛋白浓度,按照SDS-PAGE凝胶制备试剂盒制备下层胶和上层胶,蛋白上样、电泳、转膜,然后用无蛋白快速封闭液封闭15 min,一抗4 ℃冰箱孵育过夜(Nrf2、GPX4、xCT、HO-1、FSP1 抗体稀释比例均为1∶1000,β-actin 抗体稀释比例为1∶5000)。TBST洗膜3次后,二抗(稀释比例为1∶5000)室温孵育1 h,重复上述洗膜步骤,显影,使用Image J软件分析蛋白条带灰度值。
1.10 统计学方法
用GraphPad Prism9.5统计学软件进行统计分析和作图,计量资料以均数±标准差表示,组间比较采用单因素方差分析,Plt;0.05认为差异具有统计学意义。
2 结果
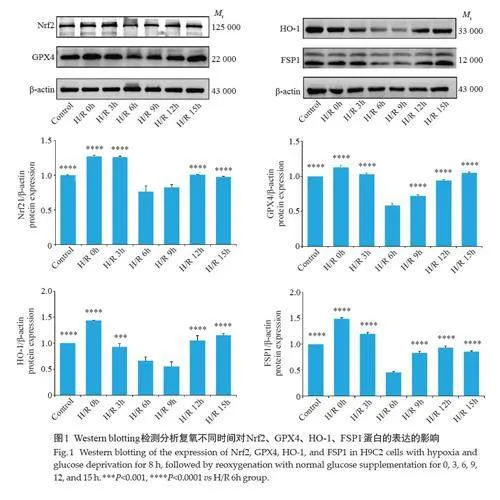
2.1 通过Western blotting检测复氧不同时间对GPX4、FSP1等蛋白表达的影响
在Nrf2、HO-1蛋白的表达中,H/R 6 h组与H/R 9 h组相比差异无统计学意义(Pgt;0.05),H/R 6 h组和除H/R 9 h组之外的其他组相比,表达量最少,差异有统计学意义(Plt;0.001);在GPX4 和FSP1 蛋白表达中,H/R 6 h组与其余各组相比蛋白表达量最少,差异有统计学意义(Plt;0.0001,图1)。
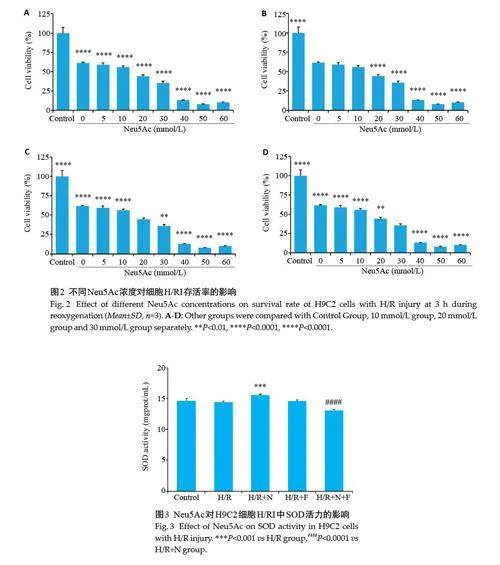
2.2 CCK-8检测不同Neu5Ac浓度对H9C2心肌细胞H/RI后的影响
与Control 组相比,实验组细胞活力均明显降低(Plt;0.0001),且随药物浓度逐渐增大,细胞活力逐渐减小;0 mmol/L组和5 mmol/L组、10 mmol/L组相比,差异均无统计学意义(Pgt;0.05),其余各组与10 mmol/L组相比,差异有统计学意义(Plt;0.0001);各组与20 mmol/L或与30 mmol/L相比,差异有统计学意义(Plt;0.01),该药物的半抑制浓度IC50= 30.07 mmol/L(图2)。
2.3 细胞SOD活性检测
Control组、H/R+F组分别与H/R组相比,SOD活力差异均无统计学意义(Pgt;0.05)。与H/R组相比,H/R+N组SOD的活力增加(Plt;0.001);H/R+N+F组SOD活力低于H/R+N组(Plt;0.0001,图3)。
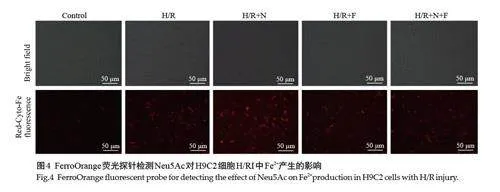
2.4 FerroOrange荧光探针检测细胞中Fe2+水平
与Control组相比较,H/R组Fe2+荧光强度增强,表明细胞内的Fe2+含量增加;与H/R组相比,H/R+N组Fe2+荧光强度增强,细胞内Fe2+含量增加;与H/R+N组相比,H/R+N+F 组Fe2+荧光强度减弱,胞内Fe2+含量减少(图4)。
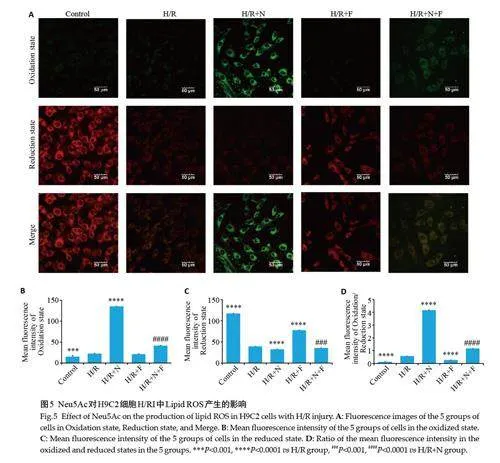
2.5 C11 BODIPY 581/591 荧光探针检测脂质过氧化水平
5 组细胞氧化态下的平均荧光强度,H/R 组高于Control组(Plt;0.001), H/R+N组高于H/R组(Plt;0.0001),H/R+F组与H/R组相比差异无统计学意义(Pgt;0.05),H/R+N+F 组荧光强度低于H/R+N组(Plt;0.0001);5 组细胞还原态下的平均荧光强度,H/R组低于Control 组和H/R+F(Plt;0.0001),H/R+N组低于H/R组(Plt;0.0001),H/R+N+F组高于H/R+N组(Plt;0.001);5组细胞氧化态的平均荧光强度与还原态的平均荧光强度的比值,Control 组和H/R+F 组低于H/R组(Plt;0.0001),H/R+N组高于H/R组(Plt;0.0001),H/R+N+F组低于H/R+N组(Plt;0.0001,图5)。
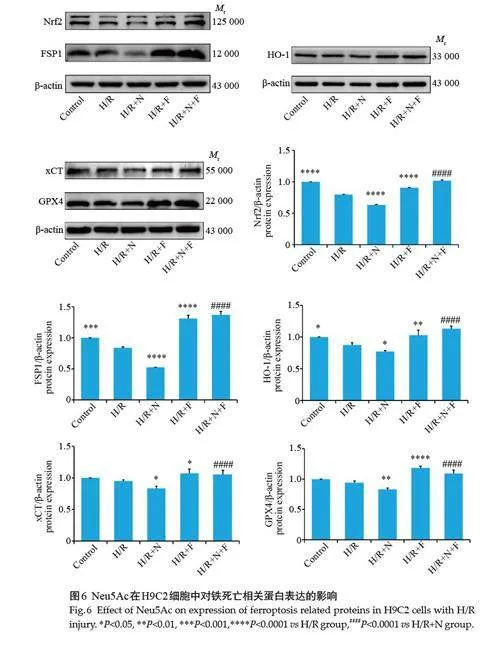
2.6 通过Western blotting检测Neu5Ac在H9C2心肌细胞H/RI中对GPX4、FSP1等蛋白表达的影响
与Control 组相比,H/R组xCT、GPX4 蛋白表达无明显差别,蛋白HO-1、Nrf2、FSP1 表达降低(Plt;0.05);与H/R 组相比,H/R+N组5 种蛋白的表达均下降(Plt;0.05);与H/R组相比,H/R+F组5种蛋白的表达均升高(Plt;0.05);与H/R+N组相比,H/R+N+F组5种蛋白的表达均升高(Plt;0.0001,图6)。
3 讨论
MIRI是心肌供血中断一段时间后血管再通恢复血液供应,原缺血的心肌功能尚不能恢复,且发生更严重损伤的一种现象,可表现为再灌注心律失常、心功能降低和心肌坏死面积增大[21]。目前认为MIRI的损伤机制与氧化应激、细胞内钙离子超载、血管内皮细胞损伤以及细胞凋亡等因素有关[22]。
铁死亡是以铁依赖性脂质过氧化为主要特征的细胞死亡方式,在肿瘤耐药、心血管疾病、神经退行性疾病、脑卒中等多种疾病或损伤的生理病理进程中发挥重要作用,是有别于细胞凋亡、细胞焦亡、细胞自噬的新型细胞程序性死亡[23]。铁死亡过程主要包括Fe2+的聚集、铁依赖的活性氧(ROS)产生、脂质过氧化和抗氧化核心酶GPX4表达的降低。大量文献报道了铁死亡在MIRI损伤中的作用,认为铁死亡是MIRI损伤的一个重要发病机制,对铁死亡的调节可能成为改善MIRI损伤的关键[24]。研究表明,Neu5Ac可通过促进冠脉粥样硬化间接损伤心肌细胞,心肌缺血再灌注患者血清中Neu5Ac明显升高[25]。本研究结果表明,Neu5Ac在MIRI中可促进心肌细胞内Fe²⁺、ROS和脂质过氧化物的积累,并下调Nrf2、GPX4、HO-1、xCT和FSP1蛋白的表达。相反,在Neu5Ac 与Fer-1 联合干预时,心肌细胞内Fe²⁺、ROS和脂质过氧化物的生成显著减少。超氧化物歧化酶(SOD)是心血管系统中清除ROS的关键抗氧化酶,其活性与细胞内ROS生成量呈正相关[26]。C11 BODIPY581/591脂质过氧化荧光探针由于其良好的亲脂性,能够快速进入细胞膜并选择性捕获细胞内及膜中的脂质过氧化物。因此,通过检测SOD活性和C11 BODIPY581/591脂质过氧化荧光探针的荧光强度,可间接反映细胞内ROS和脂质过氧化物的水平。
Nrf2 是调控氧化应激反应的关键转录因子,与细胞自我保护作用密切相关,其与抗氧化物反应元件(ARE)结合形成体内重要的抗氧化应激Nrf2/ARE通路而启动抗氧化应激反应[27]。此外研究表明,Nrf2通路的激活通过增加抗氧化酶活性,抑制过度氧化应激延缓糖尿病心肌病进展,从而可以改善心脏的缺血再灌注损伤[28]。同时Nrf2也是调控铁死亡的重要转录因子,xCT和铁蛋白是受Nrf2 调控的重要靶基因,Nrf2 的核移位上调xCT和铁蛋白表达、减轻脂质过氧化,减少铁离子蓄积,进而抑制铁死亡的发生[29]。本研究在蛋白水平观察了Neu5Ac对Nrf2 表达的影响,研究结果显示,经历缺氧复氧损伤的心肌细胞中Nrf2 的表达明显降低,同时与铁死亡相关的GPX4、HO-1、xCT和FSP1蛋白表达也显著降低。使用铁死亡抑制剂Fer-1 后,能够逆转Neu5Ac 对Nrf2、GPX4、HO-1、xCT 和FSP1 蛋白的下调,提示Neu5Ac对缺血再灌注心肌损伤作用可能与激活铁死亡有关。
综上所述,Neu5Ac 在MIRI 中可能通过抑制Nrf2信号轴的信号传导,导致活性氧和脂质活性氧的过度积累,从而加剧心肌细胞的铁死亡。目前,MIRI的长期治疗措施包括钙离子拮抗剂、β受体阻滞剂以及他汀类调脂药物的应用[30]。本研究结果表明,通过应用神经氨酸酶抑制剂减少Neu5Ac的产生,可减少心肌细胞铁死亡并改善MIRI相关并发症,从而为MIRI的防治提供了新的理论依据。此外,本研究为急性冠脉综合征的临床靶向治疗及改善预后提供了潜在的干预策略。
参考文献:
[1] 刘明波, 王增武, 樊 静, 等.《 中国心血管健康与疾病报告2023》要点
解读[J]. 中国心血管病研究, 2024, 22(7): 577-93.
[2] Szczeklik W, Fronczek J. Myocardial injury after noncardiac surgery
‑an update[J]. Curr Opin Anaesthesiol, 2021, 34(3): 381-6.
[3] Shi XJ, Li MN, Xuan L, et al. Clinical characteristics of patients with
premature acute coronary syndrome and adverse cardiovascular
events after PCI[J]. Exp Ther Med, 2019, 18(1): 793-801.
[4] Gunata M, Parlakpinar H. A review of myocardial ischaemia/
reperfusion injury: Pathophysiology, experimental models,
biomarkers, genetics and pharmacological treatment[J]. Cell
Biochem Funct, 2021, 39(2): 190-217.
[5] Chen H, Zhu J, Le YF, et al. Salidroside inhibits doxorubicininduced
cardiomyopathy by modulating a ferroptosis-dependent
pathway[J]. Phytomedicine, 2022, 99: 153964.
[6] Ding SY, Duanmu XY, Xu LS, et al. Ozone pretreatment alleviates
ischemiareperfusion injury-induced myocardial ferroptosis by
activating the Nrf2/Slc7a11/Gpx4 axis[J]. Biomed Pharmacother,
2023, 165: 115185.
[7] Qian SE, Long Y, Tan GL, et al. Programmed cell death: molecular
mechanisms, biological functions, diseases, and therapeutic targets
[J]. MedComm, 2024, 5(12): e70024.
[8] Heimerl M, Sieve I, Ricke-Hoch M, et al. Neuraminidase-1
promotes heart failure after ischemia/reperfusion injury by affecting
cardiomyocytes and invading monocytes/macrophages[J]. Basic
Res Cardiol, 2020, 115(6): 62.
[9] Varki A. Sialic acids in human health and disease[J]. Trends Mol
Med, 2008, 14(8): 351-60.
[10]Heimerl M, Gausepohl T, Mueller JH, et al. Neuraminidases-key
players in the inflammatory response after pathophysiological
cardiac stress and potential new therapeutic targets in cardiac disease
[J]. Biology, 2022, 11(8): 1229.
[11] Liu ZH, Xiang P, Zeng SM, et al. N-Acetylneuraminic acid triggers
endothelial pyroptosis and promotes atherosclerosis progression via
GLS2-mediated glutaminolysis pathway[J]. Cell Death Discov,
2024, 10(1): 467.
[12]Xiang P, Chen QQ, Chen L, et al. Metabolite Neu5Ac triggers
SLC3A2 degradation promoting vascular endothelial ferroptosis
and aggravates atherosclerosis progression in ApoE-/- mice[J].
Theranostics, 2023, 13(14): 4993-5016.
[13]Zhang L, Wei TT, Li Y, et al. Functional metabolomics characterizes
a key role for N-acetylneuraminic acid in coronary artery diseases
[J]. Circulation, 2018, 137(13): 1374-90.
[14]Gayral S, Garnotel R, Castaing-Berthou A, et al. Elastin-derived
peptides potentiate atherosclerosis through the immune Neu1-PI3Kγ
pathway[J]. Cardiovasc Res, 2014, 102(1): 118-27.
[15]Hu XM, Li YY, Chen QY, et al. Sialic acids promote macrophage
M1 polarization and atherosclerosis by upregulating ROS and
autophagy blockage[J]. Int Immunopharmacol, 2023, 120: 110410.
[16]Chen L, Qiu HM, Chen QQ, et al. N-acetylneuraminic acid
modulates SQSTM1/p62 sialyation-mediated ubiquitination
degradation contributing to vascular endothelium dysfunction in
experimental atherosclerosis mice[J]. IUBMB Life, 2024, 76(3):
161-78.
[17]Li MN, Qian SH, Yao ZY, et al. Correlation of serum NAcetylneuraminic
acid with the risk and prognosis of acute coronary
syndrome: a prospective cohort study[J]. BMC Cardiovasc Disord,
2020, 20(1): 404.
[18]Li MN, Bao BW, Ding SY, et al. Correlation between plasma
glutathione peroxidase 4 and N-acetylneuraminic acid levels with
clinical risk stratification and prognosis of patients with acute
coronary syndrome[J]. Saudi Med J, 2022, 43(10): 1103-10.
[19]Chen XC, Sun T, Qi YX, et al. Paeoniflorin ameliorates reperfusion
injury in H9C2 cells through SIRT1-PINK1/parkin-mediated
mitochondrial autophagy[J]. Mol Immunol, 2024, 177: 32-43.
[20]Gao Y, Song LL, Xu JT, et al. The role of exosomes in myocardial
ischemia-reperfusion injury[J]. Cardiology, 2024: 1-17.
[21]Shi PL, Sha YT, Wang XR, et al. Targeted delivery and ROSresponsive
release of lutein nanoassemblies inhibit myocardial
ischemia-reperfusion injury by improving mitochondrial function
[J]. Int J Nanomedicine, 2024, 19: 11973-96.
[22]Zavadovsky KV, Ryabov VV, Vyshlov EV, et al. Intra-myocardial
hemorrhage and cardiac microvascular injury in ischemia/
reperfusion. A systematic review of current evidences[J]. Curr
Probl Cardiol, 2025, 50(1): 102918.
[23]Brown AR, Hirschhorn T, Stockwell BR. Ferroptosis-disease perils
and therapeutic promise[J]. Science, 2024, 386(6724): 848-9.
[24]Zhong CN, Dong H, Ma YT, et al. Single-cell sequencing combined
with transcriptomics and invivo and invitro analysis reveals the
landscape offerroptosis in myocardial ischemia-reperfusion injury
[J]. FASEB J, 2024, 38(21): e70164.
[25]Schauer R. Sialic acids: fascinating sugars in higher animals and man
[J]. Zoology, 2004, 107(1): 49-64.
[26]He ML, Li XY, Guo YQ, et al. Nerol attenuates doxorubicin-induced
heart failure by inhibiting cardiomyocyte apoptosis in rats[J]. Eur J
Pharmacol, 2024, 987: 177203.
[27]Abdelmawgood IA, Kotb MA, Hassan HS, et al. Gentisic acid
attenuates ovalbumin-induced airway inflammation, oxidative
stress, and ferroptosis through the modulation of Nrf2/HO-1 and NF-
κB signaling pathways[J]. Int Immunopharmacol, 2024, 146:
113764.
[28]Yang F, Smith MJ. Metal profiling in coronary ischemia-reperfusion
injury: implications for KEAP1/NRF2 regulated redox signaling[J].
Free Radic Biol Med, 2024, 210: 158-71.
[29]Liu DH, Zhu YZ. Unveiling smyd-2's role in cytoplasmic nrf-2
sequestration and ferroptosis induction in hippocampal neurons after
cerebral ischemia/reperfusion[J]. Cells, 2024, 13(23): 1969.
[30] Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the
management of acute myocardial infarction in patients presenting
with ST-segment elevation: the Task Force for the management of
acute myocardial infarction in patients presenting with ST-segment
elevation of the European Society of Cardiology (ESC) [J]. Eur
Heart J, 2018, 39(2): 119-77.
(编辑:经 媛)
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:16:04
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:24
中国医药导报(2017年6期)2017-04-06 16:44:01
新教育时代·教师版(2017年2期)2017-03-23 12:04:55
中国医药导报(2016年32期)2017-02-28 17:00:36
中国现代医生(2016年23期)2016-11-15 02:47:50
海南医学(2016年8期)2016-06-08 05:43:00
中国民族民间医药·下半月(2015年12期)2016-02-25 15:00:25
中国病理生理杂志(2015年8期)2015-12-21 12:38:08
医学信息(2015年15期)2015-07-07 14:59:00
