
药物的长效化设计策略
2025-02-06 00:00:00黄慕齐蔡铮刘叔文
南方医科大学学报 2025年1期
摘要:随着药物设计和制剂技术的进步,长效药物的开发已成为精准医疗与慢病管理的重要研究方向。此类药物通过延长体内有效浓度的维持时间,减少用药频率达到改善患者依从性与生活质量的目的。小分子药物、单克隆抗体及核酸药物在实现长效化方面各有难点,特别是后两者因其结构的复杂性,存在更多挑战。本文将对小分子药物、单克隆抗体及核酸药物的长效化设计策略进行综述。
关键词:长效药物;药物设计;小分子药物;单克隆抗体;核酸药物
长效药物是指通过特定的分子设计或制剂手段,使药物在机体内保持相对稳定、持续的有效浓度,从而显著减少给药频次的药物类型。与常规药物相比,此类药物通常具有更长的生物半衰期或以缓控释方式维持有效药物水平,给药周期可从数日或数周延长至数月,甚至数年。对于需长期用药的患者来说,持续稳定的药物疗效可降低症状波动,提高生活质量,并减少因频繁给药导致的依从性问题。未来,随着精准医学的发展,长效药物将在更多领域展现潜力,包括神经退行性疾病、免疫调控、罕见病治疗及肿瘤维持治疗等方面。
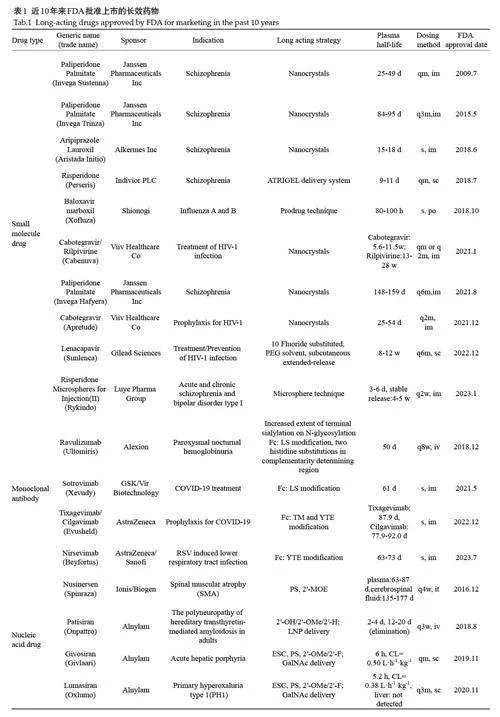
近10 年来FDA批产上市的长效药物见表1,不同类型药物的长效设计策略不同。小分子药物主要依靠前药设计、缓控释技术、及植入式给药平台实现长效。单克隆抗体(简称单抗)通过工程化改造Fc区、优化糖基化模式、聚乙二醇(PEG)修饰及纳米载体递送,有效延长其血液循环时间并降低清除率。核酸药物则借助化学修饰与特定配体共轭或脂质纳米粒等递送载体,提高在体内的稳定性与特异性,使给药间隔显著延长。本文将从小分子药物、单抗和核酸药物的长效化设计角度,综述各自的研究进展,以期为长效药物的研发提供参考。
1 小分子药物长效化设计策略
小分子药物结构相对简单,但多在体内被快速代谢与清除,需通过特定的制剂学策略延长给药间隔。目前小分子药物主要借助前药设计、微晶技术、微球技术及改变给药途径等多重手段进行设计。
1.1 前药设计
前药技术将药物(母药)与脂肪酸长链(如癸酸、对映酸或己酸)共价结合,形成亲脂性前药,并溶于特定的油性基质(如芝麻油、蓖麻油)中。酯类前体药物不仅增加其在油相中的溶解度,还增强其在脂肪组织中的分布。前药在体内可以通过酯键水解释放母药,分布到脂肪组织的前药又逐渐向体内缓慢释放前药,补充到酯键水解的循环中,最终实现药物的缓释[1]。这类技术常用于长效小分子药物开发,比如第一代典型抗精神病药类长效药物如癸酸氟奋乃嗪(Lyogen Depot)和氟哌啶醇癸酸酯(Fluanxol Depot)等。氟哌啶醇癸酸酯是氟哌啶醇与癸酸结合形成的酯类化合物,肌注后从肌肉组织的油剂中缓慢、持久、稳定地释放至组织和血液及淋巴循环中,经水解酶催化水解后,游离的氟哌啶醇进入血液,作用时间比氟哌啶醇长9~20倍,生物半衰期达到3周,每4周重复注射1次,3个月达稳态浓度[2]。
1.2 微晶技术
微晶技术通过对药物颗粒的微米化处理,并结合悬浮制剂技术,使药物在体内逐步释放,从而实现缓释或长效治疗效果[3]。棕榈酸帕利哌酮(PaliperidonePalmitate)是早期抗精神病药物利培酮(Risperidone)的体内主要活性代谢产物帕利哌酮(Paliperidone)的棕榈酸前药。棕榈酸帕利哌酮注射液1个月制剂(PP1M,商品名:Invega Sustenna)、3 个月制剂(PP3M,商品名:Invega Trinza)和6 个月制剂(PP6M,商品名:InvegaHafyera)制剂工艺相似,都是活性成份帕利哌酮转化为疏水性的帕利哌酮棕榈酸酯,再利用纳米晶体技术制成微粒悬浮液。肌肉注射后,这些微粒在肌肉组织中逐渐释放药物,并被体内酯酶缓慢水解为活性药物帕利哌酮,实现长效释放。通过适当增加微粒大小和药物浓度,进一步延长药物的缓释时间,3种药物的生物半衰期分别达到25~49 d、84~95 d 和148~159 d,分别实现每月、每3个月和每半年给药一次的长效治疗效果,大幅提高了治疗依从性[4]。HIV整合酶抑制剂Cabotegravir(卡博特韦)口服片剂的平均血浆半衰期较短,约为31.5 h,利用纳米晶体技术制成长效纳米悬浮剂,显著延长了血浆半衰期至25~54 d,肌内注射或皮下注射后52周仍可在血液中保持着相当的浓度[5]。卡博特韦注射液或者其两药复方卡博特韦/利匹韦林注射液,每年仅需给药6次或12次,极大地改善患者的治疗体验和便利性[6, 7]。
1.3 微球技术
药物递送领域的载药微球,是指将药物溶解或分散于聚合物材料中所形成的微小球体或类球体,粒径一般在1~250 μm范围内。药物经过微球技术处理后,被包封在聚合物内的药物不易被释放,聚合物在生理环境下缓慢溶蚀降解,使得包载的药物在体内以一定速率缓慢释放,从而实现药物长时间地(从几天到几个月)释放和治疗,代表药物如注射用奥曲肽微球(SandostatinLAR)、注射用利培酮微球(Risperdal Consta)和纳曲酮缓释微球(Naltrexone)等[8-10]。值得一提的是,绿叶制药集团自主创新研发的微球制剂注射用利培酮微球(Ⅱ),通过高载药量低突释微球制备技术,改善了药物释放周期和释放速度。相比于Risperdal Consta在首次注射后约3周的释药延滞期,注射用利培酮微球(Ⅱ)首次注射后,开始有少量初始释放,随后可稳定持续释放4~5周,利培酮及其主要活性代谢产物9-羟基利培酮的中位达峰时间均为17 d,无药物迟滞期,因此无需同时补充口服制剂,提高临床治疗过程的便利性[11]。
1.4 改变给药途径
肠外制剂可以避免药物的首过效应,并且可以通过制剂技术精确控制药物的释放速度,从而在延长药物作用时间,减少给药次数。很多肠外长效制剂如微球制剂、长效水性纳米/微混悬液和脂质体注射剂等,通过皮下或肌肉注射可以调整药物释放速率,延长作用时间至数周甚至数月。Lenacapavir(来那卡帕韦)作为一种首创的长效HIV-1衣壳抑制剂,其长效性得益于其在化学结构优化过程中通过引入吸电子基团(如10个氟原子)和增加分子刚性策略,增强其肝微粒体稳定性。放射性标记人体物质平衡研究结果显示[12],Lenacapavir 单次静脉给药后,在健康受试者的血浆中原形药物占总血浆暴露量的69%,其代谢产物均小于总血浆暴露量的10%;原形药物在粪便中的总放射性含量约76%,在尿液中的总放射性含量小于1%,表明Lenacapavir主要通过肠道排泄。Lenacapavir口服给药的中位半衰期为10~12 d,而其缓释制剂使用聚乙二醇300作为溶剂,皮下注射后在注射部位形成药物储库,有一个缓慢吸收和释放的过程,中位半衰期延长至8~12周,进而实现一年给药两次的长效性[13]。该研究成果被Science杂志评为2024年年度十大科学突破之首,在艾滋病治疗和预防方面取得了显著成功,有望帮助结束艾滋病的流行[14]。
2 单克隆抗体药物长效化设计策略
单抗的长效化不同于小分子,核心原则在于延缓蛋白清除和提升在体内循环的生物半衰期。IgG类抗体在哺乳动物体内的清除受新生儿Fc受体(FcRn)介导的再循环机制调控。通过优化Fc区域,使抗体在低pH环境(如内体条件)下与FcRn的结合更紧密,可减少溶酶体降解并将抗体释放回循环系统,实现生物半衰期延长。这类优化往往结合微调糖基化和氨基酸突变,以减少非特异性清除与聚集,还可利用高分子修饰、酶切位点重组以及融合蛋白策略增强分子稳定性[15]。
2.1 结构修饰
单抗实现长效化策略包括对FcRn 结合特性的Fc片段工程化改造、糖基化修饰模式的精确调控以及与PEG或类似高分子链段的共价偶联。通过对Fc区域高度保守的N-糖基化位点末端唾液酸化,可以延长生物半衰期[16, 17]。聚乙二醇化技术通过聚乙二醇共价偶联抗体,降低肾脏清除率,聚乙二醇生成的水化层可以保护生物大分子免受酶降解,显著延长药物在体内的滞留时间[18, 19]。目前实现单抗长效化的核心在于对FcRn结合特性的Fc片段工程化。FcRn负责维持IgG和白蛋白的循环以及跨极化细胞屏障的双向运输,以pH依赖性方式与IgG结合,尤其在酸性pH值(pH 6.0)下能紧密结合。此外,FcRn和Fc之间的疏水相互作用通过FcRn上的阴离子残基与Fc区的质子化组氨酸或谷氨酸残基之间形成的盐桥来稳定。因此通过在FcRn-Fc界面处对Fc区残基进行诱变,可以增强这种相互作用,从而延长IgG的生物半衰期[20]。
通过工程化Fc区域以延长其生物半衰期的多种单抗药物已进入临床使用。Sotrovimab 是一种由VirBiotechnology 与葛兰素史克(GSK)合作开发的针对SARS-CoV-2病毒的单抗。通过在Fc区域进行M428L和N434S 氨基酸取代(LS 修饰),增加了与FcRn 的结合,其群体药代动力学数据显示中位生物半衰期为61 d[21]。使用LS修饰实现长效化的还有Ravulizumab,一种长效的抗C5单抗,通过对原始药物Eculizumab(增加N-糖基化上的末端唾液酸化程度)进行结构改造,首先在互补决定区域进行两个组氨酸替换,其次在Fc区进行LS修饰,提高了与FcRn的结合亲和力,尤其是pH6.0时的亲和力,消除了靶向介导的抗体清除。通过这种改造,Ravulizumab 的生物半衰期达到了50 d,相比Eculizumab延长了4倍,患者只需每8周接受一次静脉注射治疗,大大减少了维持剂量的频率[22,23]。
另一种常用Fc 片段工程化方法为YTE 修饰,即M252Y/S254T/T256E位点的三重氨基酸突变,增加了与FcRn 的结合,可促进单抗血清生物半衰期增加四倍[24]。Tixagevimab 和Cilgavimab 在TM修饰(L234F/L235/P331S取代)的基础上使用YTE修饰,延长了生物半衰期[25]。Nirsevimab(尼塞韦单抗)是一种靶向呼吸道合胞病毒(RSV)F蛋白融合前构象(pre-F)的全人源重组IgG1κ单抗,2024年1月被中国NMPA批准上市,用于预防新生儿和婴儿由RSV引起的下呼吸道感染。Nirsevimab通过在Fc片段上引入YTE修饰,可减少抗体的降解,将生物半衰期延长至63~73 d,接种一剂次即可提供长达5个月的保护效果[26, 27]。
Fc 片段的工程化设计不仅用于单抗,还被广泛应用于双特异性抗体和融合蛋白等新型抗体结构的开发,进一步提高了这些分子的稳定性和药代动力学特性。
2.2 载体递送
药物递送系统可以维持药物在血液中的浓度,从而使药物可以继续作用更长的时间。目前,可用的抗体药物控释系统包括水凝胶、微球和植入物等。水凝胶具有高吸水能力的交联亲水聚合物三维网状结构,物理限制药物释放,以及其生物粘附特性延长药物在特定部位的保留时间,从而减少药物清除速率,延长药物在体内的作用时间。Bevacizumab(贝伐珠单抗)利用水凝胶为载体,药物可缓慢释放约30 d,证明水凝胶是玻璃体内注射微创治疗年龄相关性黄斑变性(AMD)的一种极具潜力的载体[28]。除水凝胶外,微球也已成为治疗AMD的新策略。由PLGA(聚乳酸-羟基乙酸共聚物)和PEGPLA(聚乙二醇-聚乳酸)共聚物构成的微球制剂,能够高效地包载贝伐珠单抗,并且能够持续释放贝伐珠单抗长达90 d[29]。目前,越来越多的新型植入物已被用于抗体递送,有研究人员制备了固体脂质植入物(SLIs),将Ranibizumab和IgG1类的单抗纳入脂质体基质中,观察到这两种抗体药物能够持续释放大约120 d[30]。
3 核酸药物长效化设计策略
核酸药物通过特异性识别并结合内源性核酸序列,调控基因表达或影响RNA功能,从根源上抑制或修正致病基因表达,成为精准医疗中的重要工具。由于核酸分子在体内易被核酸酶降解,且分子量和带电性较高,递送和稳定性是实现长效的首要难点。提升分子内在稳定性、降低免疫系统识别以及增强组织特异性摄取,对于延长核酸药物的生物半衰期和生物学效应时间至关重要。与小分子缓控释不同,核酸药物往往通过特定的化学修饰(如磷酸骨架和核糖修饰)和递送系统优化使核酸在体内得以持续存在并保持有效功能[31]。
3.1 磷酸骨架修饰
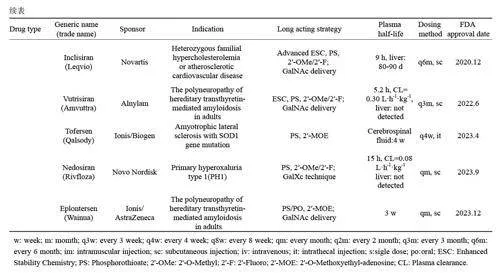
磷酸酯键是核酸结构中的关键连接点,但天然的磷酸二酯键易被体内的核酸酶降解。硫原子替换磷酸基团上的一个非桥连氧原子被广泛应用于反义寡核苷酸(ASO)和小干扰核酸(siRNA)中,这一修饰不仅可以显著提高核酸药物的抗酶解能力,还能促进与血清、细胞表面和细胞内的蛋白质结合,这些相互作用可以增加细胞摄取和组织分布,从而延长药物在体内的生物半衰期[32]。硫代磷酸酯键(PS)修饰是提高siRNA 5'和3'端稳定性的主要策略。Alnylam 公司开发的新一代siRNA使用了增强稳定化学(ESC)修饰方式,用硫代磷酸酯键取代反义链3'和5'端以及正义链5'端的两个末端磷酸二酯键,提高了代谢稳定性,从而减少了所需的总剂量和给药频率[33]。Givosiran是首款采用ESC设计的N-乙酰半乳糖胺(GalNAc)偶联siRNA,2019年被FDA批准用于治疗急性肝卟啉症(AHP),其皮下注射给药的推荐剂量为每月2.5 mg/kg,有效减少了给药频率。Inclisiran 则采用更高级的ESC设计,在推荐的300 mg剂量下,观察到药物在ASCVD(动脉粥样硬化性心血管疾病)或HeFH(家族性高胆固醇血症)受试者肝脏中的生物半衰期约80~90 d,每6个月重复皮下注射1次,能在数月内(超过6个月)持续降低LDL-C水平[34]。
3.2 核糖修饰
核糖修饰涉及对RNA分子的核糖部分进行结构改变,2'位置的修饰对于防止核酸被核酸酶降解至关重要,最常用的修饰包括2'-O-甲基(2'-OMe)、2'-甲氧基乙氧基(2'-MOE)和2'-氟(2'-F)修饰。2'-O-Me和2'-F修饰通过保护2'-羟基(2'-OH)免受核酸酶水解,增强了siRNA的稳定性[35, 36]。6 款已上市的siRNA药物都采用2'-OMe和2'-F修饰以延续药物生物半衰期,实现数月给药一次的长效治疗。2'-甲氧乙氧基修饰(2'-MOE)作为2'-甲氧基的类似物,在结合目标mRNA和抵抗酶解方面表现更为出色,被广泛应用于ASO药物[37]。Nusinersen是目前唯一一种在中国获批上市并被纳入医保的ASO药物,用于治疗脊髓性肌萎缩症。Nusinersen的每个核苷酸都采用了2'-MOE修饰,大多数的碱基进行了5 号位的甲基化修饰,相比与最原始的ASO,在血浆中的平均终末生物半衰期63~87 d,及在脑脊液(CSF)的平均终末生物半衰期为135~177 d,仅需每4 个月进行一次注射治疗,为患者减少许多痛苦[38]。
3.3 载体递送
由于核酸类药物的高分子量和负电荷性,难以穿透细胞膜,导致组织的递送受限和细胞摄取不良。因此,开发有效的递送技术对实现高效的细胞摄取、内体逃逸和药物在细胞内的释放具有重大意义。目前,用于核酸药物递送的各种载体处于不同的开发阶段,包括脂质纳米颗粒(LNP)、GalNAc、细胞穿透肽和内源性外泌体等。目前商业化最成功的递送载体之一就是LNP,LNP主要由可电离的阳离子脂质、PEG修饰脂质、胆固醇和天然磷脂组成。可电离的阳离子脂质是LNP成功开发的核心所在,可以调控mRNA进入细胞后的内含体逃逸过程,PEG 修饰脂质能够在空间上形成一个包裹mRNA的亲水层,阻碍单核巨噬细胞的摄取和血浆蛋白的结合[39]。目前已有3 款使用LNP 的核酸药物获批,Patisiran是首款LNP-siRNA,其临床药代动力学数据显示[40],1次/周静脉注射0.3 mg/kg,ALN-18328(siRNA分子)和DLin-MC3-DMA(阳离子脂质)的血浆PK曲线相似,出现初始阶段的快速消除和次级峰及相对较长的末端消除半衰期,ALN-18328在稳态下的t1/2β为2~4 d,预估97%的ALN-18328将在给药后约12~20 d消除,多次给药应无蓄积,但稳态时血浆AUC的累积比为2~3倍,归因于siRNA与阳离子脂质的结合,有很大部分LNP从晚期内体/溶酶体流出回到循环中,又被肝脏重新分配和消除。在新冠疫情期间,Pfizer/BioNTech 和Moderna 公司开发了两款mRNA疫苗Tozinameran 和Elasomeran,均利用LNP作为递送系统,实现间隔3 周或4 周接种2 剂次可有效预防COVID-19,这些疫苗的成功应用展示了LNP-mRNA技术在传染病预防领域的重要价值[41, 42]。
GalNAc是目前应用最广泛的递送系统,目前已上市和临床试验中的siRNA药物绝大多数使用GalNAc偶联技术。相对于LNP等其他药物递送技术,GalNAc结构也更为简单,成本较低,量产更为方便。研究表明,内体逃逸是GalNAc 偶联物活性的限速步骤,GalNAc偶联物通过去唾液酸糖蛋白受体(ASGPR)介导的内吞途径运输并积累在酸性细胞内区室(如溶酶体)中,从这些隔室中缓慢释放siRNA或ASO,从而延长药物的作用时间[43, 44]。同样是治疗转甲状腺素蛋白淀粉样变性多发性神经病(针对TTR 靶点)的两款ASO 药物,Eplontersen和Inotersen的用法用量分别为45 mg/月和300 mg/周,两者在化学修饰上基本相似,而Eplontersen采用GalNAc递送系统。根据说明书信息可知,Inotersen进入体内后,很快被代谢,而Eplontersen在血浆中几乎不被代谢,进入肝脏后8~72 h代谢释放ASO,导致作用时间长于Inotersen,药物生物半衰期约3周[45, 46]。
4 总结与展望
长效药物的开发已在小分子药物、单抗和核酸药物等领域取得了重要进展,小分子药物通过前药设计、微晶和微球技术等延长药物的释放速度和释放时间,单抗依赖Fc改造和聚合物纳米载体等延缓蛋白清除速率,延长在体内循环的生物半衰期,核酸药物则通过化学修饰和递送载体提高药物在体内的稳定性和持久性。尽管长效药物的设计在减少用药负担和提高治疗效果方面展现了巨大价值,但仍面临剂量释放精准控制、体外与体内释放行为相关性预测、临床适用性评估及质量控制等多方面的挑战,这些均涉及药物设计、制剂工艺及临床应用的多学科交叉领域。未来,人工智能辅助设计、高效递送载体和新型生物材料的快速发展将进一步提升长效药物的精准性和安全性,不同类型药物的协同开发和技术整合有望优化药物生物半衰期、稳定性和靶向性,从而推动长效药物成为慢病管理与精准医疗的重要支柱,为患者提供更加安全、便捷且可持续的治疗选择。
参考文献:
[1] Fralish Z, Chen A, Khan S, et al. The landscape of small-molecule
prodrugs[J]. Nat Rev Drug Discov, 2024, 23(5): 365-80.
[2] 张鸿燕, 舒 良, 周东丰, 等. 氟哌啶醇的药代动力学研究[J]. 中华神
经精神科杂志, 1995, 28(6): 325-8.
[3] Kalhapure RS, Palekar S, Patel K, et al. Nanocrystals for controlled
delivery: state of the art and approved drug products[J]. Expert
Opin Drug Deliv, 2022, 19(10): 1303-16.
[4] Peters L, Dyer M, Schroeder E, et al. Invega hafyera (paliperidone
palmitate): extended-release injectable suspension for patients with
schizophrenia[J]. J Pharm Technol, 2023, 39(2): 88-94.
[5] Landovitz RJ, Li SE, Eron JJ Jr, et al. Tail-phase safety, tolerability,
and pharmacokinetics of long-acting injectable cabotegravir in HIVuninfected
adults: a secondary analysis of the HPTN 077 trial[J].
Lancet HIV, 2020, 7(7): e472-e481.
[6] Zeuli JD, Rivera CG, Smith BL, et al. Cabotegravir: a novel HIV
integrase inhibitor combined with rilpivirine as the first long-acting
injectable program for the treatment of HIV infection[J]. Drugs
Today, 2022, 58(12): 555-76.
[7] Hodge D, Back DJ, Gibbons S, et al. Pharmacokinetics and drugdrug
interactions of long-acting intramuscular cabotegravir and
rilpivirine[J]. Clin Pharmacokinet, 2021, 60(7): 835-53.
[8] Su Y, Zhang BL, Sun RW, et al. PLGA-based biodegradable
microspheres in drug delivery: recent advances in research and
application[J]. Drug Deliv, 2021, 28(1): 1397-418.
[9] Markowicz-Piasecka M, Kubisiak M, Asendrych-Wicik K, et al.
Long-acting injectable antipsychotics-a review on formulation and
in vitro dissolution[J]. Pharmaceutics, 2023, 16(1): 28.
[10]Correll CU, Kim E, Sliwa JK, et al. Pharmacokinetic characteristics
of long-acting injectable antipsychotics for schizophrenia: an
overview[J]. CNS Drugs, 2021, 35(1): 39-59.
[11]中华医学会精神医学分会精神分裂症协作组. 注射用利培酮微球临
床应用专家共识[J]. 中国心理卫生杂志, 2023, 37(8): 641-7.
[12]Weber E, Subramanian R, Rowe W, et al. Pharmacokinetics,
disposition, and biotransformation of[14C]lenacapavir, a novel, firstin-
class, selective inhibitor of HIV-1 capsid function, in healthy
participants following a single intravenous infusion[J]. Clin
Pharmacokinet, 2024, 63(2): 241-53.
[13]Di Perri G. Pharmacological outlook of Lenacapavir: a novel first-inclass
Long-Acting HIV-1 Capsid Inhibitor[J]. Infez Med, 2023, 31
(4): 495-9.
[14]Kelley CF, Acevedo-Quiñones M, Agwu AL, et al. Twice-yearly
lenacapavir for HIV prevention in men and gender-diverse persons
[J]. N Engl J Med, 2024,[Online ahead of print].
[15]Wang H, Song MD, Xu JQ, et al. Long-acting strategies for antibody
drugs: structural modification, controlling release, and changing the
administration route[J]. Eur J Drug Metab Pharmacokinet, 2024, 49
(3): 295-316.
[16]Bas M, Terrier A, Jacque E, et al. Fc sialylation prolongs serum halflife
of therapeutic antibodies[J]. J Immunol, 2019, 202(5): 1582-94.
[17]Boune S, Hu PS, Epstein AL, et al. Principles of N-linked
glycosylation variations of IgG-based therapeutics: pharmacokinetic
and functional considerations[J]. Antibodies, 2020, 9(2): 22.
[18]Reichard EE, Nanaware-Kharade N, Gonzalez GA 3rd, et al.
PEGylation of a high-affinity anti-(+)methamphetamine single chain
antibody fragment extends functional half-life by reducing clearance
[J]. Pharm Res, 2016, 33(12): 2954-66.
[19]Shi YJ, Lu A, Wang XY, et al. A review of existing strategies for
designing long-acting parenteral formulations: focus on underlying
mechanisms, and future perspectives[J]. Acta Pharm Sin B, 2021, 11
(8): 2396-415.
[20]Lee CH, Kang TH, Godon O, et al. An engineered human Fc domain
that behaves like a pH-toggle switch for ultra-long circulation
persistence[J]. Nat Commun, 2019, 10(1): 5031.
[21]Gupta A, Gonzalez-Rojas Y, Juarez E, et al. Early treatment for
covid-19 with SARS-CoV-2 neutralizing antibody sotrovimab[J]. N
Engl J Med, 2021, 385(21): 1941-50.
[22]Rondeau E, Scully M, Ariceta G, et al. The long-acting C5 inhibitor,
Ravulizumab, is effective and safe in adult patients with atypical
hemolytic uremic syndrome naïve to complement inhibitor treatment
[J]. Kidney Int, 2020, 97(6): 1287-96.
[23]McKeage K. Ravulizumab: first global approval[J]. Drugs, 2019, 79
(3): 347-52.
[24]Dall' Acqua WF, Kiener PA, Wu H. Properties of human IgG1s
engineered for enhanced binding to the neonatal Fc receptor (FcRn)
[J]. J Biol Chem, 2006, 281(33): 23514-24.
[25]Loo YM, McTamney PM, Arends RH, et al. The SARS-CoV-2
monoclonal antibody combination, AZD7442, is protective in
nonhuman Primates and has an extended half-life in humans[J]. Sci
Transl Med, 2022, 14(635): eabl8124.
[26]Griffin MP, Yuan Y, Takas T, et al. Single-dose nirsevimab for
prevention of RSV in preterm infants[J]. N Engl J Med, 2020, 383
(5): 415-25.
[27]Hammitt LL, Dagan R, Yuan Y, et al. Nirsevimab for prevention of
RSV in healthy late-preterm and term infants[J]. N Engl J Med,
2022, 386(9): 837-46.
[28]Hu CC, Chiu YC, Chaw JR, et al. Thermo-responsive hydrogel as an
anti-VEGF drug delivery system to inhibit retinal angiogenesis in
Rex rabbits[J]. Technol Health Care, 2019, 27(S1): 153-63.
[29] Iyer S, Radwan AE, Hafezi-Moghadam A, et al. Long-acting
intraocular Delivery strategies for biological therapy of age-related
macular degeneration[J]. J Control Release, 2019, 296: 140-9.
[30]Vollrath M, Engert J, Winter G. Long-term release and stability of
pharmaceutical proteins delivered from solid lipid implants[J]. Eur
J Pharm Biopharm, 2017, 117: 244-55.
[31]Crooke ST, Witztum JL, Bennett CF, et al. RNA-targeted
therapeutics[J]. Cell Metab, 2018, 27(4): 714-39.
[32]Yu RZ, Lemonidis KM, Graham MJ, et al. Cross-species comparison
of in vivo PK/PD relationships for second-generation antisense
oligonucleotides targeting apolipoprotein B-100[J]. Biochem
Pharmacol, 2009, 77(5): 910-9.
[33]Nair JK, Attarwala H, Sehgal A, et al. Impact of enhanced metabolic
stability on pharmacokinetics and pharmacodynamics of GalNAcsiRNA
conjugates[J]. Nucleic Acids Res, 2017, 45(19): 10969-77.
[34]Gosselin NH, Schuck VJA, Barriere O, et al. Translational
population-pharmacodynamic modeling of a novel long-acting
siRNA therapy, inclisiran, for the treatment of hypercholesterolemia
[J]. Clin Pharmacol Ther, 2023, 113(2): 328-38.
[35]Podbevsek P, Allerson CR, Bhat B, et al. Solution-state structure of a
fully alternately 2'-F/2'-OMe modified 42-nt dimeric siRNA
construct[J]. Nucleic Acids Res, 2010, 38(20): 7298-307.
[36]Sheng L, Rigo F, Bennett CF, et al. Comparison of the efficacy of
MOE and PMO modifications of systemic antisense oligonucleotides
in a severe SMA mouse model[J]. Nucleic Acids Res, 2020,
48(6): 2853-65.
[37]Crooke ST, Baker BF, Crooke RM, et al. Antisense technology: an
overview and prospectus[J]. Nat Rev Drug Discov, 2021, 20(6):
427-53.
[38]Claborn MK, Stevens DL, Walker CK, et al. Nusinersen: a treatment
for spinal muscular atrophy[J]. Ann Pharmacother, 2019, 53(1):
61-9.
[39]Eygeris Y, Gupta M, Kim J, et al. Chemistry of lipid nanoparticles
for RNA delivery[J]. Acc Chem Res, 2022, 55(1): 2-12.
[40]Zhang XP, Goel V, Robbie GJ. Pharmacokinetics of patisiran, the
first approved RNA interference therapy in patients with hereditary
transthyretin-mediated amyloidosis[J]. J Clin Pharmacol, 2020, 60
(5): 573-85.
[41] Jung HN, Lee SY, Lee S, et al. Lipid nanoparticles for delivery of
RNA therapeutics: current status and the role of in vivo imaging[J].
Theranostics, 2022, 12(17): 7509-31.
[42]Hald Albertsen C, Kulkarni JA, Witzigmann D, et al. The role of
lipid components in lipid nanoparticles for vaccines and gene therapy
[J]. Adv Drug Deliv Rev, 2022, 188: 114416.
[43]Brown CR, Gupta S, Qin JE, et al. Investigating the
pharmacodynamic durability of GalNAc-siRNA conjugates[J].
Nucleic Acids Res, 2020, 48(21): 11827-44.
[44]Prakash TP, Graham MJ, Yu JH, et al. Targeted delivery of antisense
oligonucleotides to hepatocytes using triantennary N-acetyl
galactosamine improves potency 10-fold in mice[J]. Nucleic Acids
Res, 2014, 42(13): 8796-807.
[45]Yu RZ, Collins JW, Hall S, et al. Population pharmacokineticpharmacodynamic
modeling of inotersen, an antisense
oligonucleotide for treatment of patients with hereditary
transthyretin amyloidosis[J]. Nucleic Acid Ther, 2020, 30(3):
153-63.
[46]Nie TN. Eplontersen: first approval[J]. Drugs, 2024, 84(4): 473-8.
(编辑:经 媛)
