
UHPLC-MS/MS法同时测定调味品中5种脂溶性色素和5种罂粟壳生物碱的残留量
2024-12-31 00:00:00丁文慧田洪霞朱伟伟张帅
中国调味品 2024年10期




摘要:建立了一种经QuEChERS EMR(Enhanced Matrix Removal)-Lipid净化结合超高效液相色谱-串联质谱(UHPLC-MS/MS)同时测定调味品中苏丹红Ⅰ~Ⅳ、对位红5种脂溶性色素和罂粟碱、吗啡、可待因、蒂巴因、那可丁5种罂粟壳生物碱残留量的高通量分析方法。罂粟碱、蒂巴因、那可丁和5种脂溶性色素在1~100 ng/mL范围内线性关系良好,相关系数均大于0.99,定量限(LOQ)为1.0 μg/kg;吗啡、可待因在8~100 ng/mL范围内线性关系良好,相关系数均大于0.99,定量限(LOQ)为5.0 μg/kg;各化合物平均回收率为81.2%~104.5%,相对标准偏差为2.9%~11.4%(n=6)。该方法简便、灵敏、稳定、通量高、成本低,适用于香辛料调味油、火锅底料及蘸料、麻辣烫底料等多种调味品的检测质控需求。
关键词:QuEChERS EMR-Lipid;超高效液相色谱-串联质谱;脂溶性色素;罂粟壳生物碱;调味品
中图分类号:TS201.2""""" 文献标志码:A"""" 文章编号:1000-9973(2024)10-0179-07
Simultaneous Determination of Residual Amount of Five Lipid Soluble Pigments
and Five Poppy Husk Alkaloids in Seasonings by UHPLC-MS/MS Method
DING Wen-hui1, TIAN Hong-xia1, ZHU Wei-wei1, ZHANG Shuai2
(1.School of Food and Drug, Weifang Vocational College, Weifang 261000, China;
2.Anqiu Inspection and Testing Center Co., Ltd., Weifang 262100, China)
Abstract: A high-throughput analysis method is established for the simultaneous determination of residual amount of five lipid soluble pigments such as Sudan Red Ⅰ~Ⅳ and Para Red, as well as five poppy husk alkaloids such as papaverine, morphine, codeine, thebaine and narcotine in seasonings using QuEChERS EMR (Enhanced Matrix Removal)-Lipid purification combined with ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). Papaverine, thebaine, narcotine and five lipid soluble pigments show a good linear relationship within the range of 1~100 ng/mL, the correlation coefficients are all greater than 0.99, and the limit of quantification (LOQ) is 1.0 μg/kg.Morphine and codeine show a good linear relationship within the range of 8~100 ng/mL, the correlation coefficients are both greater than 0.99, and the limit of quantification (LOQ) is 5.0 μg/kg. The average recovery rates of each compound range from 81.2% to 104.5%, with the relative standard deviations between 2.9% and 11.4% (n=6).The method is simple, sensitive, stable, high-throughput and low-cost, and it is suitable for the detection and quality control requirements of various seasonings such as" spice flavored oil, hotpot seasoning and dipping sauce, and Malatang seasoning.
Key words: QuEChERS EMR-Lipid; UHPLC-MS/MS; lipid soluble pigments; poppy husk alkaloids; seasoning
苏丹红Ⅰ~Ⅳ、对位红是脂溶性偶氮类工业染料,禁止用于食品染色。苏丹红及其代谢产物具有致癌性、遗传毒性、致敏性;对位红对眼睛、皮肤和呼吸系统有刺激性,其毒性尚不明确。由于此类染料廉价易得、着色稳定、添加后产品鲜艳光亮,故仍有不法商家在调味品中使用。罂粟壳生物碱属于非食用违禁物质,长期食用会对人体神经系统、消化系统造成损害。根据整顿办函[2011]1号和食品整治办[2008]3号文件的要求:食品中不得检出苏丹红Ⅰ~Ⅳ和罂粟壳生物碱成分。因此,建立一种先进且通用易行的检测方法是十分必要的。
苏丹红Ⅰ~Ⅳ、对位红的检测方法有薄层色谱法[1]、液相色谱法 [2-4]和液相色谱-串联质谱法 [5-6];罂粟壳生物碱的检测方法有气相色谱-质谱法[7]、液相色谱-串联质谱法[8-10]。薄层色谱法重现性差且缺少有效的定性确认手段;液相色谱-串联质谱法较液相色谱法具有更强的抗干扰能力,定性准确、灵敏度高;较气相色谱-质谱法具有更高的重现性和准确性。目前,能够同时测定脂溶性色素和罂粟壳生物碱残留量的高通量检测方法尚未见报道。
本研究采用增强型脂质去除吸附剂(Enhanced Matrix Removal-Lipid,EMR-Lipid)QuEChERS净化方法,一次预处理同时检测脂溶性色素和罂粟壳生物碱残留量,通过UHPLC-MS/MS进行定性定量分析,方法高效准确,为调味品中非法违禁添加物的检测和质量安全评价提供了技术支持。
1 材料与方法
1.1 仪器与试剂
1290Ⅱ高效液相色谱仪、6470三重四极杆质谱仪(配鞘流电喷雾离子源AJS ESI,MassHunter质谱工作站) 美国Agilent公司;ME240E分析天平 瑞士梅特勒-托利多公司;HT-200多管旋涡混合仪 浙江赛德仪器设备有限公司;3K15冷冻离心机 德国Sigma公司;AS-E40UVT-R纯水仪 重庆奥思德仪器设备有限公司。
乙腈、乙酸铵、甲酸(均为色谱纯):德国CNW公司;EMR-Lipid、EMR-Lipid Polish、陶瓷均质子:美国Agilent公司;乙酸(分析纯):国药集团化学试剂有限公司;苏丹红Ⅰ(99.0%)、苏丹红Ⅱ(99.0%)、苏丹红Ⅲ(96.0%)、苏丹红Ⅳ(94.0%)、对位红(99.9%)、苏丹红Ⅰ-D5(98.0%)、苏丹红Ⅲ-D6(98.0%)、苏丹红Ⅳ-D6(98.0%):德国Dr.公司;吗啡(100%)、吗啡-D3(100%)、可待因-D3(100%):美国Cerilliant公司;罂粟碱(99.9%)、可待因(100%)、蒂巴因(100%)、那可丁(99.6%):英国政府化学家实验室;苏丹红Ⅱ-D6(98.0%):印度Clearsynth公司;实验室用超纯水。
1.2 标准溶液的配制
分别准确称取标准品及内标于10 mL容量瓶中,使用乙腈溶解并定容,制备1 mg/mL标准储备液。吸取各标准储备液,分别制备1 μg/mL混合标准工作液及混合内标标准工作液。
1.3 样品的制备
将粉状及液体调味品混合均匀后,密封、标记,于4 ℃冷藏存放;固体及半固体调味品经粉碎后混合均匀,密封、标记,于4 ℃冷藏存放。
1.4 样品的前处理
1.4.1 提取
准确称量5.0 g(精确至0.01 g)样品于50 mL具塞离心管中,加入混合内标标准工作液150 μL。粉状、固体及半固体样品需提前加入5 mL H2O超声处理5 min。加入2粒陶瓷均质子、15 mL 0.2%甲酸乙腈溶液,涡旋5 min,超声5 min,再次涡旋5 min,9 500 r/min离心1 min,待净化。
1.4.2 净化
向EMR-Lipid净化管中加入5 mL H2O,快速振摇2 min。移取10 mL提取液至净化管中,涡旋2 min,5 000 r/min离心1 min,移取全部上清液至EMR-Lipid Polish反萃管中,涡旋1 min,5 000 r/min离心1 min。移取5 mL上层有机相于10 mL玻璃管中,于40 ℃氮吹至干,加入1 mL 50%乙腈,复溶后过0.22 μm尼龙滤膜,待测定。
1.4.3 标准加入法绘制标准曲线
称取6份空白样品于50 mL具塞离心管中,分别加入混合标准工作液3,30,60,150,240,300 μL,加入混合内标标准工作液150 μL。参照1.4.1、1.4.2中前处理方法获得质量浓度分别为1,10,20,50,80,100 ng/mL的基质匹配标准曲线,其内标浓度为50 ng/mL。
1.5 实验条件
1.5.1 色谱条件
色谱柱:ZORBAX Eclipse Plus C18色谱柱(3.0 mm×50 mm,1.8 μm);流动相:A为5 mmol/L乙酸铵水溶液(含0.1%甲酸),B为乙腈;流速:0.5 mL/min;梯度洗脱程序:0~1.00 min,5% B;1.00~3.50 min,5%~80% B;3.50~3.60 min,80%~98% B;3.60~6.00 min,98% B;6.00~6.01 min,98%~5% B;6.01~7.00 min,5% B;柱温:40 ℃;进样量:1 μL。
1.5.2 质谱条件
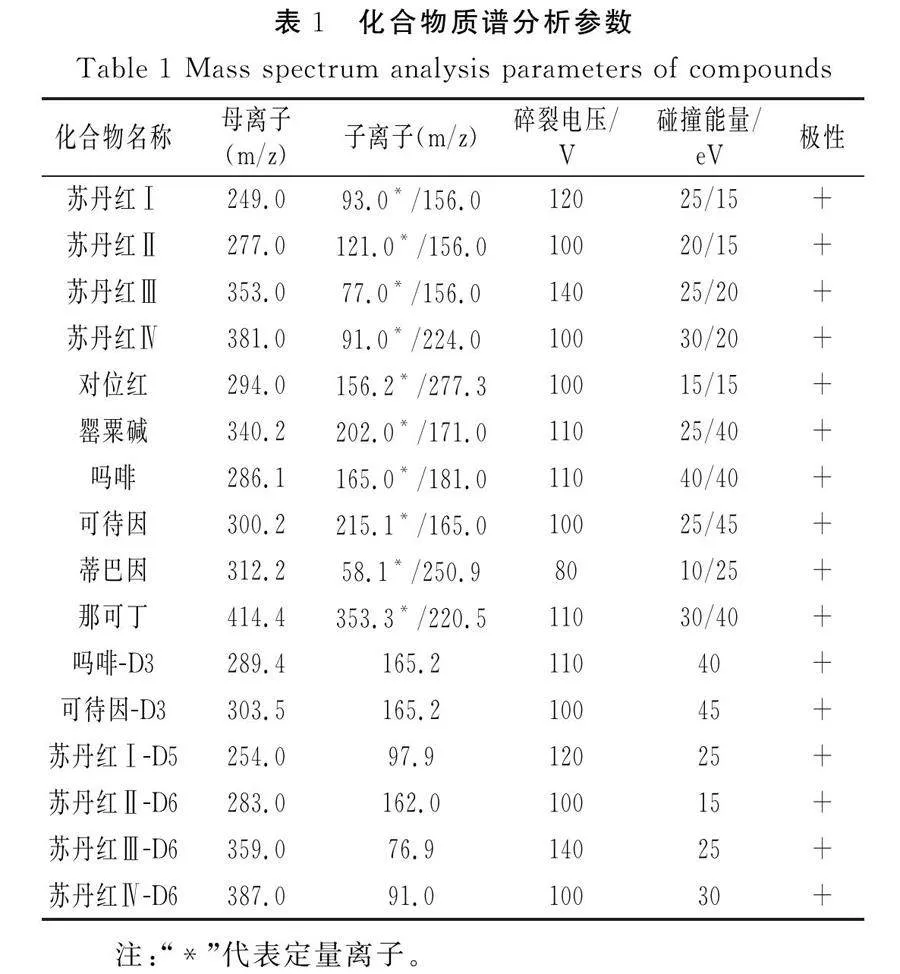
采用三重四极杆质谱仪;检测模式:正离子动态多反应监测(+DMRM);干燥气温度325 ℃,干燥气流速7 L/min;鞘气温度300 ℃,鞘气流速10 L/min;雾化器压力276 kPa;毛细管电压3 000 V;喷嘴电压0 V;Delta EMV(+)400;干燥气:氮气;碰撞气:高纯氮气。各化合物定性定量离子对、碎裂电压、碰撞能量及极性见表1。
2 结果与讨论
2.1 前处理条件优化
2.1.1 提取方式的选择
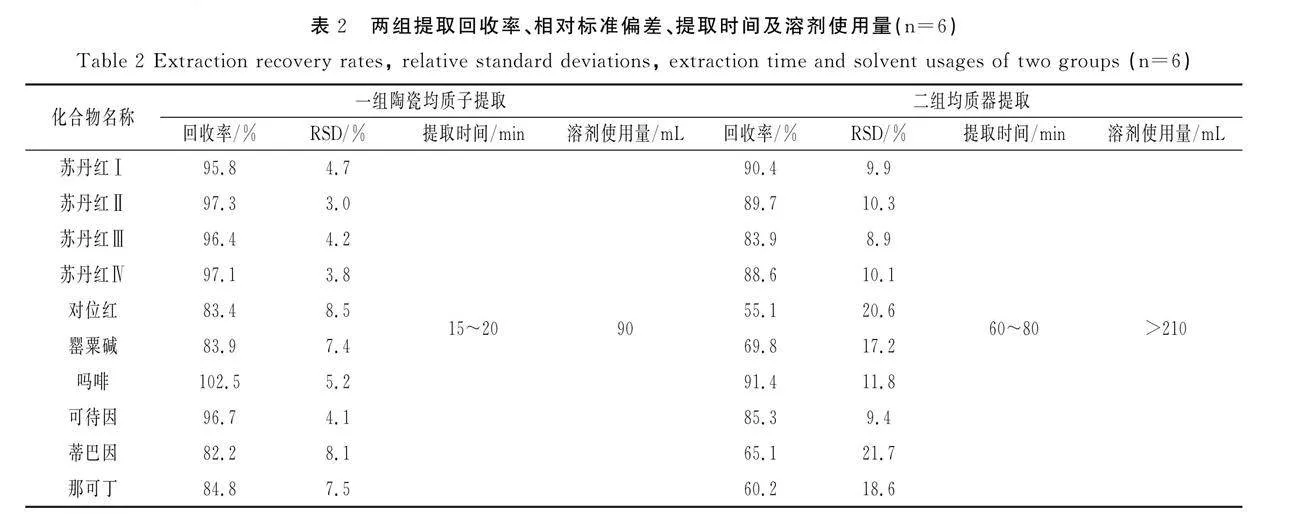
含有动物油脂的火锅底料、部分香辛料调味油十分黏稠,制样冷藏后易结块,样品在振荡过程中易成球状,涡旋提取不充分。目前,多采用均质器均质提取的方式,但均质后需用提取液清洗刀头并合并提取液。多个样品同时进行前处理时,为避免交叉污染,需对刀头充分清洗,增加了工作量和试剂使用量[11]。本研究将陶瓷均质子(经惰性化处理,对目标化合物无吸附)置于离心管中,代替均质器均质,并对两种方式进行比较。选取制样均匀的牛油火锅底料,取样12份,加入等量混合标准工作液200 μL,涡旋混合均匀后冷藏保存48 h。将其分成两组:一组采用陶瓷均质子提取,二组采用均质器提取。提取回收率、相对标准偏差、提取时间及溶剂使用量见表2。结果表明,一组回收率为82.2%~102.5%,相对标准偏差为3.0%~8.5%;二组回收率为55.1%~91.4%,相对标准偏差为8.9%~21.7%。陶瓷均质子提取提高了萃取效率且溶剂使用量少、提取过程简单,可最大程度地减少技术人员操作误差,具有较好的重复性。因此,本研究选用陶瓷均质子分散样品。
2.1.2 净化方式的选择与基质效应
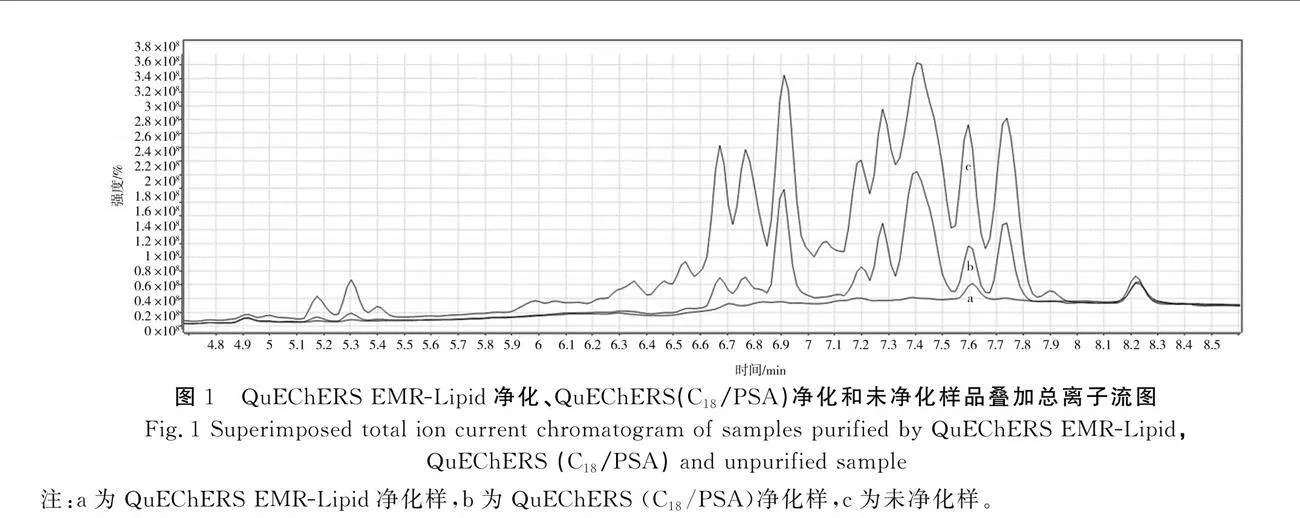
调味品中含有大量的油脂和多种色素,为降低基质干扰且充分发挥仪器性能,需对样品进行净化。目前,样品净化多采用QuEChERS(C18/PSA)法[7],该法成本低、效率高、通量高、操作简便,可实现多类别不同化合物的同时净化[11],但此法对颜色深、高脂类样品基质的净化效果不理想。本研究比较了QuEChERS EMR-Lipid净化、QuEChERS(C18/PSA)净化和未净化样品提取液的LC-MS/MS全扫描谱图,其基质净化效果见图1。由图1可知,经QuEChERS EMR-Lipid净化后离子流图背景洁净度高,降低了基质干扰,提高了方法的灵敏度和积分的准确性。通过对全扫描谱图积分,计算得出QuEChERS EMR-Lipid和QuEChERS(C18/PSA)的基质去除率分别为94.9%和50.4%。因此,本研究选用QuEChERS EMR-Lipid法对样品进行净化。
根据斜率法[12]基质效应公式ME(%)=(基质标曲的斜率/溶剂标曲的斜率-1)×100%,若|ME|<20%,属弱基质效应;若20%≤|ME|≤50%,属中等基质效应;若|ME|>50%,属强基质效应[11]。本研究对质量浓度为1~100 ng/mL的基质标曲与溶剂标曲分别进样,根据上述公式得苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、对位红、罂粟碱、吗啡、可待因、蒂巴因、那可丁的|ME|值分别为13.2%、8.8%、9.8%、11.4%、19.7%、11.7%、14.2%、7.5%、12.6%、10.1%,均小于20%,属弱基质效应,说明净化效果较好。
2.1.3 提取溶剂的选择
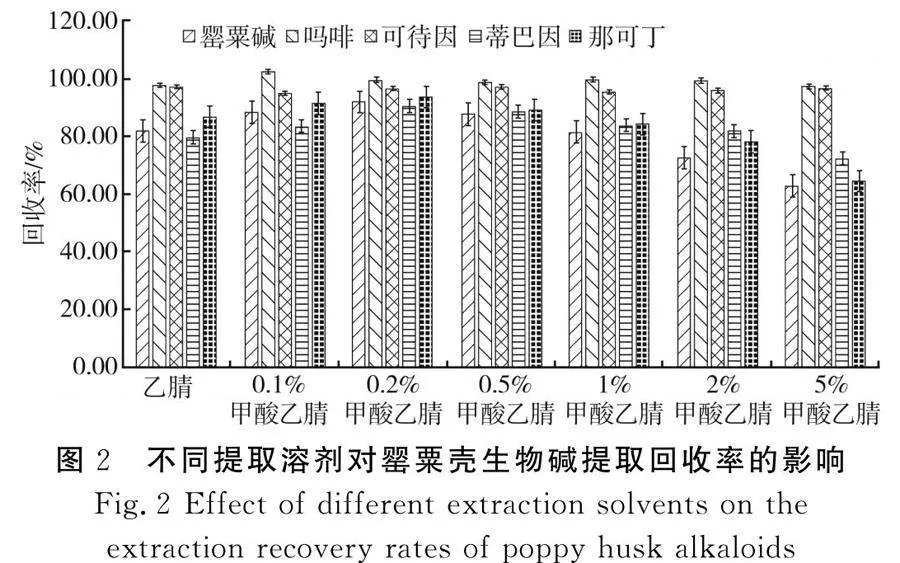
苏丹红Ⅰ~Ⅳ、对位红易溶于正己烷[3-4]、乙腈[5-6,9]、丙酮[13]等有机溶剂;罂粟壳生物碱可通过乙腈[9]、甲酸乙腈[8]或0.1 mol/L盐酸[10]提取。乙腈可兼容两类化合物,在乙腈中添加甲酸能够促进目标化合物与样品解离,改善提取效果。本研究比较了乙腈、0.1%甲酸乙腈、0.2%甲酸乙腈、0.5%甲酸乙腈、1%甲酸乙腈、2%甲酸乙腈、5%甲酸乙腈的回收率。随着甲酸浓度的增加,苏丹红Ⅰ~Ⅳ、对位红的回收率没有明显改变,维持在85.6%~102.3%。罂粟碱、吗啡、可待因、蒂巴因、那可丁的回收率见图2。
由图2可知,因吗啡、可待因可通过内标校正,回收率较稳定;而罂粟碱、蒂巴因、那可丁在甲酸含量为0.2%时回收率最高,此后随着甲酸浓度的升高而降低,这与甲酸导致化合物离子化趋势增大有关,使一部分目标物被分配至水相中。因此,本研究选择0.2%甲酸乙腈作为提取溶剂。
2.1.4 复溶溶剂的选择
样品经盐析后直接以乙腈相进样,样液的洗脱强度明显高于初始流动相。进入色谱柱后,扩散到流动相中的目标物以正常速度被洗脱,而样液中的目标物因强溶剂洗脱导致移动速度快、目标物峰形扭曲、峰展宽或峰分裂。为进一步抑制溶剂效应,本研究采用分取浓缩并复溶的方式,对比5%、10%、20%、30%、40%、50%、60%、70%乙腈水溶液作为复溶溶剂的回收率及溶剂效应对峰形的影响。
由图3中a可知,当乙腈比例小于40%时,苏丹红Ⅰ~Ⅳ、对位红的溶解性差,回收率偏低;当乙腈比例大于60%时,因强溶剂比例较高,出峰较早的吗啡出现溶剂效应,出现严重峰分裂,难以定性定量。由图3中b可知,当乙腈比例为50%时,可有效抑制柱外扩散,目标物峰形细长,无峰分裂。且当乙腈比例为50%时,目标物的溶解性好,回收率均高于80%。因此,本研究对提取液适当浓缩后选用50%的乙腈水溶液作为复溶溶剂。
2.2 色谱条件优化
2.2.1 色谱柱的选择
本研究比较了Kinetex HILIC色谱柱(2.1 mm×100 mm,1.7 μm)、ZORBAX Eclipse Plus C18色谱柱(3.0 mm×50 mm,1.8 μm)、ZORBAX SB-Aq色谱柱(4.6 mm×150 mm,5 μm)的保留行为[14]。结果表明,苏丹红Ⅰ~Ⅳ、对位红在HILIC色谱柱上无保留,这与苏丹红类药物分子中氢原子易与相邻氮原子形成分子内氢键而极性较弱有关;在SB-Aq色谱柱上能够获得较强的分离与保留,但填料粒径过大且色谱柱较长,限制了超高效液相性能的发挥,18 min左右完成进样;而在Plus C18色谱柱上各化合物分离及保留适中,峰形尖锐,7 min左右完成进样。因此,本研究选用Plus C18色谱柱。
2.2.2 流动相的选择及梯度设定
本研究以乙腈为流动相(有机相部分),比较了0.1%甲酸水溶液和5 mmol/L乙酸铵水溶液(含0.1%甲酸)作为流动相(水相部分)时目标物的分离效果和响应值。结果表明,当流动相中未添加乙酸铵时,目标物的分离度及峰形较差;当添加乙酸铵后,减少了次级保留,目标物的分离度及峰形得到明显改善,提高了响应值。因此,本研究选用乙腈-5 mmol/L乙酸铵水溶液(含0.1%甲酸)作为流动相。
因两类化合物的极性差别较大,故采用梯度洗脱方式。在1.00 min内,设置95%的水相并缓慢增加有机相比例至3.50 min,使强极性的吗啡保留时间在色谱柱的死时间外,保证准确定量;当罂粟壳生物碱被完全洗脱后,迅速提高有机相比例至98%并恒流2.4 min,使弱极性的脂溶性色素被快速洗脱并获得良好的分离效果。在此条件下各化合物的色谱图见图4。
2.3 质谱条件优化
在正离子模式下通过全扫描获得各化合物分子离子峰,以此为母离子优化碎裂电压;随后进行选择离子扫描,选取丰度较高、干扰较小的离子为定量和定性离子;最后在多反应监测模式下优化各子离子的碰撞电压[15]。为进一步提高化合物的响应强度,按鞘气温度、鞘气流速、喷嘴电压、毛细管电压、雾化器压力、干燥器温度、干燥器流速的顺序进一步优化相关参数,优化后的质谱参数见表1。将各参数设成标准MRM采集方式并在优化好的色谱条件下进样,利用设备软件自带的Update DMRM Method功能转换成DMRM(动态多反应监测)采集模式。DMRM采集方式只在化合物窗口宽度内扫描,可有效减少同时采集的离子对数目并在一定程度上增加了驻留时间,使每种化合物的色谱峰所采集的点数不少于20~30个,保证定量的准确性。因此,本研究选用DMRM采集方式,设置每条离子对的保留时间和窗口宽度[11],各化合物的保留时间由标准MRM获取并可在方法转换时自动识别;窗口宽度通常为1 min。
2.4 方法学验证
2.4.1 线性关系和定量限
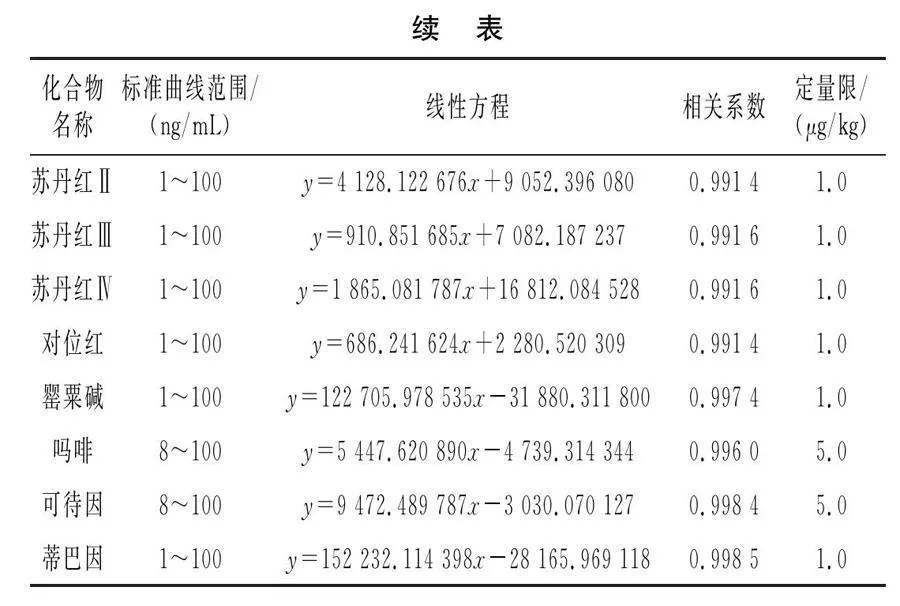
对质量浓度为1,8,10,20,50,80,100 ng/mL的基质匹配标准曲线进行测定,由MassHunter Quantitative(B.07.00)数据处理系统完成标准曲线的绘制,以各标曲点质量浓度(ng/mL)为横坐标,以定量离子峰面积为纵坐标,得到线性回归方程。对阴性样品加标,以S/N≥10、精密度≤21%、回收率60%~120%为依据,得到罂粟碱、蒂巴因、那可丁和5种脂溶性色素的方法定量限为1.0 μg/kg;吗啡、可待因的方法定量限为5.0 μg/kg,本方法能够满足残留检测需求。各化合物标准曲线范围、线性方程、相关系数(r)及定量限见表3。在1~100 ng/mL或8~100 ng/mL范围内,r均大于0.99,符合方法确证要求。
2.4.2 回收率和精密度
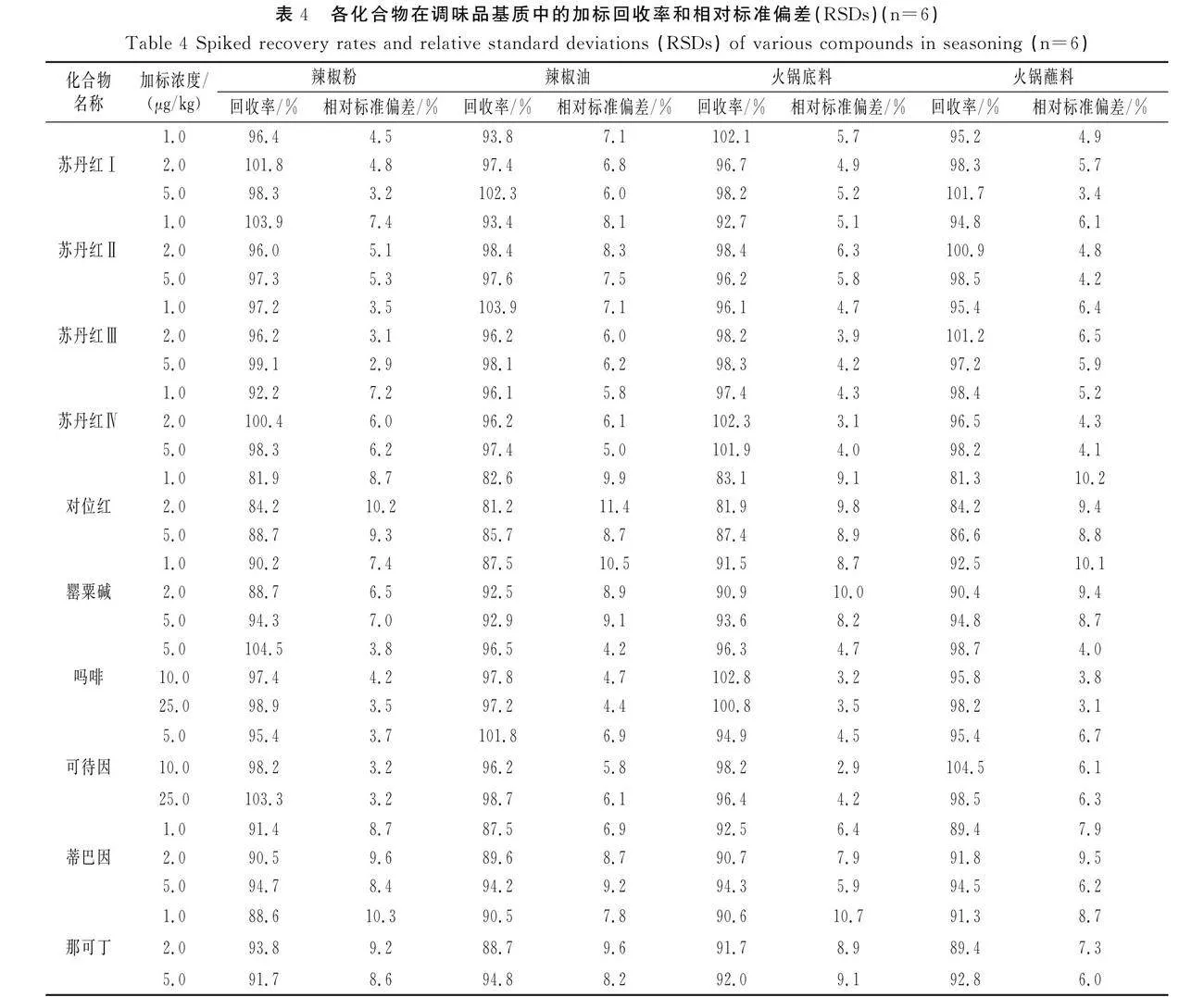
分别向空白辣椒粉、辣椒油、火锅底料、火锅蘸料中添加1倍、2倍、5倍定量限标准品,进行低、中、高三水平添加回收试验,平行测定6次,方法的回收率和精密度见表4。方法的平均回收率为81.2%~104.5%,相对标准偏差为2.9%~11.4%,方法的准确度和精密度均符合残留分析要求。
2.5 实际样品分析
对从山东省各大超市及农贸市场随机采购的60批调味品用本研究方法进行检测,样品包括10批火锅底料、10批火锅蘸料、10批辣椒粉、10批辣椒油、10批辣椒酱、10批香辛料酱,其中1批火锅底料检出吗啡3.6 μg/kg,未到定量限,其余项目均未检出;检测过程中按2倍定量限加标质控,回收率在80%~110%之间,保证定量结果的准确性。
3 结论
本研究建立了一种增强型脂质去除净化技术结合超高效液相色谱-串联质谱法同时测定5种脂溶性色素和5种罂粟壳生物碱的检验方法。该方法采用QuEChERS EMR-Lipid净化,可获得与固相萃取相当的净化效果,基质去除率达94.9%。该方法前处理简单快速、回收率高、精密度好,为调味品中非法违禁添加物质的检测和质量安全评价提供了技术支持。
参考文献:
[1]冯华,王样培,王世俊,等.辣椒粉中非法添加苏丹红色素快速检验分析[J].食品安全质量检测学报,2018,9(13):3421-3426.
[2]胡思怡,薛昆鹏,周勇,等.改进分子印迹固相萃取——高效液相色谱法同时测定辣椒制品中4种苏丹红含量[J].食品安全质量检测学报,2019,10(2):494-499.
[3]苗翠.高效液相色谱法测定禽蛋中苏丹红残留量[J].现代食品,2018(16):124-126,141.
[4]朱浩,李小平,邹宝波,等.分散固相萃取/高效液相色谱法测定鸡蛋中对位红与苏丹红染料残留[J].分析测试学报,2013,32(11):1379-1383.
[5]刘俊,朱吕,陆春燕,等.超高效液相色谱串联质谱法同时检测禽蛋中的角黄素、对位红和苏丹红Ⅰ~Ⅳ[J].食品安全质量检测学报,2018,9(18):4953-4958.
[6]张艳侠,尹丽丽,薛霞,等.超高效液相色谱-串联质谱法测定食品中柑橘红2号和苏丹红Ⅰ~Ⅳ染料[J].食品工业科技,2018,39(23):286-292.
[7]彭文浩.QuEChERs-气质联用法测定火锅汤料中的罂粟碱和吗啡[J].海峡预防医学杂志,2019,25(4):60-62.
[8]宁焕焱,黄秀丽,赖政炀,等.超高效液相色谱-串联质谱法快速测定调味料中5种罂粟壳生物碱残留量[J].中国酿造,2019,38(11):190-193.
[9]张子臣,杜国辉,曹宁,等.超高效液相-串联质谱法测定食品中罂粟壳成分[J].食品安全质量检测学报,2019,10(23):8111-8114.
[10]花露,叶平,陆杰,等.超高效液相色谱-串联质谱法同时测定辣条中的5种罂粟碱[J].中国调味品,2018,43(8):142-146.
[11]刘新辉,王情情,张瑗,等.Oasis PRiME HLB净化法测定动物源性食品中的兽药多残留[J].农产品质量与安全,2019(5):15-20.
[12]KITTLAUS S, SCHIMANKE J, KEMPE G, et al. Assessment of sample cleanup and matrix effects in the pesticide residue analysis of foods using postcolumn infusion in liquid chromatography-tandem mass spectrometry[J].Journal of Chromatography A,2011,1218(46):8399-8410.
[13]王永姣,孙晓,刘静,等.辣椒粉中非法添加苏丹红的测定方法研究[J].西北药学杂志,2017,32(6):683-686.
[14]国家质量监督检验检疫总局,国家标准化管理委员会.食品中苏丹红染料的检测方法 高效液相色谱法:GB/T 19681—2005[S].北京:中国标准出版社,2005.
[15]魏莉莉,薛霞,刘艳明,等.亲水作用色谱-串联质谱法测定蜂蜜中链霉素和双氢链霉素残留[J].色谱,2019,37(7):735-741.
收稿日期:2024-04-08
基金项目:山东省重大科技创新工程项目(2019JZZY011014);潍坊市科技发展计划项目(2021GX020)
作者简介:丁文慧(1988—),女,讲师,博士,研究方向:食品加工与安全、教育管理学。
猜你喜欢
家庭医药(2021年11期)2021-11-20 22:34:57
家庭医药(2021年21期)2021-10-21 10:19:55
食品安全导刊(2020年14期)2020-12-04 20:19:39
现代食品(2018年16期)2018-11-02 02:33:52
中国蜂业(2018年4期)2018-05-09 06:25:08
健康博览(2017年12期)2018-02-06 21:30:06
食品工业科技(2016年17期)2016-10-31 02:45:08
公民与法治(2016年4期)2016-05-17 04:09:28
当代化工研究(2016年6期)2016-03-20 16:21:46
无机化学学报(2014年3期)2014-02-28 17:30:58
