
右美托咪定通过激活Nrf2/HO-1/GPX4 通路抑制肾小管上皮细胞的铁死亡
2024-11-03 00:00:00张方圆刘刚
南方医科大学学报 2024年6期

摘要:目的 探讨右美托咪定(DEX)对Erastin诱导的人肾小管上皮细胞(HK-2)铁死亡的保护作用及其机制。方法 构建Erastin诱导的HK-2 细胞铁死亡模型。将HK-2 细胞分为对照组、Erastin 模型组、Erastin+2.5 μmol/L DEX组、Erastin+5 μmol/L DEX组、Erastin+10 μmol/L DEX组,采用CCK-8法检测细胞的存活率。将HK-2细胞分为对照组 、Erastin模型组、Erastin+10 μmol/LDEX组、Erastin+10 μmol/L DEX+ML385(Nrf2 抑制剂)组。采用CCK-8 法检测细胞的存活率;使用细胞亚铁比色法测试盒检测细胞内Fe2+水平的变化;应用流式细胞术检测细胞内活性氧(ROS)的含量;使用丙二醛(MDA)和还原型谷胱甘肽(GSH)试剂盒检测细胞中MDA及GSH含量;Western blotting 检测细胞中核因子E2 相关因子2(Nrf2)、血红素加氧酶-1(HO-1)、谷胱甘肽过氧化物酶 4(GPX4)蛋白的表达。结果 与对照组相比,Erastin 组的细胞存活率显著被抑制(Plt;0.001),同时细胞中GSH含量降低(Plt;0.001),Fe2+、ROS以及MDA含量升高(Plt;0.001);与Erastin组相比,Erastin+10 μmol/L DEX组的细胞存活率明显升高(Plt;0.001),10 μmol/L DEX显著升高细胞中GSH含量(Plt;0.001),显著降低Fe2+、ROS以及MDA含量(Plt;0.001),并且显著上调细胞中Nrf2、HO-1和GPX4蛋白的表达(Plt;0.001)。使用ML385后,Nrf2/HO-1/GPX4通路被抑制(Plt;0.001),细胞存活率降低(Plt;0.001),GSH含量降低(Plt;0.001),而Fe2+、ROS以及MDA含量升高(Plt;0.05)。结论 DEX对Erastin 诱导的HK-2 细胞铁死亡具有保护作用,其机制可能是通过激活Nrf2/HO-1/GPX4通路实现抗氧化应激从而抑制铁死亡。
关键词:右美托咪定;Erastin;人肾小管上皮细胞;铁死亡;Nrf2/HO-1/GPX4
肾小管上皮细胞铁死亡发生于急性肾损伤、肾缺血/再灌注损伤、糖尿病肾病和肾脏纤维化等多种肾脏疾病的病程中[1-4],因此靶向调控铁死亡的策略将为防治上述肾脏疾病提供新的思路。铁死亡是一种新型的细胞程序性死亡方式,是由于依赖铁离子的脂质过氧化物的过量产生而发生的[5, 6],其主要特征为细胞内铁离子聚积、活性氧(ROS)增多和谷胱甘肽(GSH)耗竭[7,8]。核因子E2相关因子2(Nrf2)是细胞抗氧化的重要核转录因子,在氧化应激条件下易位到细胞核,激活下游的血红素加氧酶-1(HO-1)和谷胱甘肽过氧化物酶4(GPX4),通过影响脂质过氧化的生成进而调控铁死亡[9-11]。
右美托咪定(DEX)是一种临床麻醉辅助用药,具有镇静、抗焦虑和镇痛等作用。近年来研究发现,DEX可通过多种途径发挥肾脏保护作用[12-14]。研究证实,DEX可通过抑制氧化应激和细胞凋亡来减轻脂多糖(LPS)诱导的人肾小管上皮细胞(HK-2)损伤[15]。然而,对于DEX确切的肾脏保护作用机制还不够清楚。DEX的肾脏保护作用与HK-2细胞铁死亡之间是否存在关联尚未有相关研究。因此,本研究将聚焦DEX对HK-2细胞铁死亡的作用及其调控机制,通过构建Erastin 诱导的HK-2细胞铁死亡模型,观察DEX对HK-2细胞铁死亡的保护作用,并以Nrf2/HO-1/GPX4抗氧化途径为切入点探讨其作用机制,为临床上肾脏疾病的治疗提供新的思路和靶点。
1 材料和方法
1.1 材料
1.1.1 细胞
人肾皮质近曲小管上皮细胞HK-2(武汉赛维尔生物科技有限公司)。
1.1.2 主要试剂
盐酸右美托咪定注射液(扬子江药业集团有限公司);Erastin、ML385(MCE);细胞亚铁比色法测试盒(Elabscience);活性氧检测试剂盒(碧云天);丙二醛检测试剂盒(赛维尔);还原型谷胱甘肽测定试剂盒(南京建成生物工程研究所);Nrf2、HO-1、β-actin 抗体(碧云天);GPX4抗体(Proteintech)。
1.1.3 主要仪器
高速冷冻离心机(艾本德);二氧化碳培养箱(PHCbi);酶标仪(Bio Tek);流式细胞仪(BD);化学发光凝胶成像系统(上海天能公司)。
1.2 方法
1.2.1 细胞培养
HK-2 细胞培养在含10% FBS 的MEM培养基中,置于37 ℃、5% CO2的细胞培养箱中,细胞达到80%融合时进行细胞传代,用于后续实验。
1.2.2 构建HK-2 细胞铁死亡模型
HK-2 细胞按6000/孔接种于96孔板中,置于37 ℃、5% CO2 细胞培养箱中培养24 h,待细胞完全贴壁后,向每孔加入铁死亡诱导剂Erastin 终浓度为2.5、5、7.5、10 μmol/L的培养基,每组设置3 个复孔。处理24 h 后,弃去旧培养基,每孔加入含10% CCK-8的培养基,37 ℃培养箱中孵育2 h后使用酶标仪检测各孔450 nm处吸光度,计算各组细胞存活率,以确定Erastin最佳的造模浓度。
1.2.3 实验分组
将HK-2细胞分为5组:对照组、Erastin模型组、Erastin+2.5 μmol/L DEX组、Erastin+5 μmol/LDEX组、Erastin+10 μmol/L DEX组,DEX预处理2 h,再加入5 μmol/L Erastin 诱导24 h 后进行后续实验;再将HK-2细胞分为4组:对照组、Erastin模型组、Erastin+10 μmol/L DEX 组、Erastin+10 μmol/L DEX+ML385组,用2 μmol/L ML385 预处理1 h,10 μmol/L DEX预处理2 h,再加入5 μmol/L Erastin诱导24 h后进行后续实验。
1.2.4 CCK8 法检测细胞存活率
HK-2 细胞接种于96孔板中,分组给药处理后,每孔加入含10% CCK-8的培养基,37 ℃培养箱中孵育2 h后使用酶标仪检测450 nm处吸光度,计算各组细胞存活率。
1.2.5 细胞内Fe2+的检测
HK-2 细胞接种于6 孔板中,分组给药处理后,消化并收集细胞,按照细胞亚铁比色法测试盒说明书中的操作于593 nm处检测吸光度,计算各组细胞内Fe2+的含量。
1.2.6 细胞内ROS的检测
HK-2细胞接种于6孔板中,分组给药处理后,加入DCFH-DA荧光探针,在37 ℃培养箱中孵育20 min后,弃旧培养液,消化并收集细胞,随后用300 μL PBS重悬,最后将重悬液通过流式细胞仪检测,并收集数据。
1.2.7 细胞内MDA、GSH的检测
细胞接种于6 孔板中,分组给药处理后,消化并收集细胞,按照MDA、GSH检测试剂盒说明书中的操作于532 nm、405 nm处检测吸光度,计算各组细胞内MDA、GSH的含量。
1.2.8 Western blotting 法检测蛋白表达
细胞接种于6孔板中,分组给药处理后,消化并收集细胞,使用裂解液提取细胞蛋白并定量,蛋白经高温变性后进行电泳、转膜、封闭、4 ℃孵育一抗过夜(Nrf2、HO-1、GPX4、β-actin抗体稀释比例均为1∶2000),洗膜后室温孵育二抗2 h,再次洗膜,随后进行化学发光显影,并采用ImageJ软件分析各组蛋白的灰度值。
1.3 统计学分析
实验数据采用GraphPad Prism8 软件进行统计分析,计量资料以均数±标准差表示,组间比较采用单因素方差分析,Plt;0.05为差异有统计学意义。
2 结果
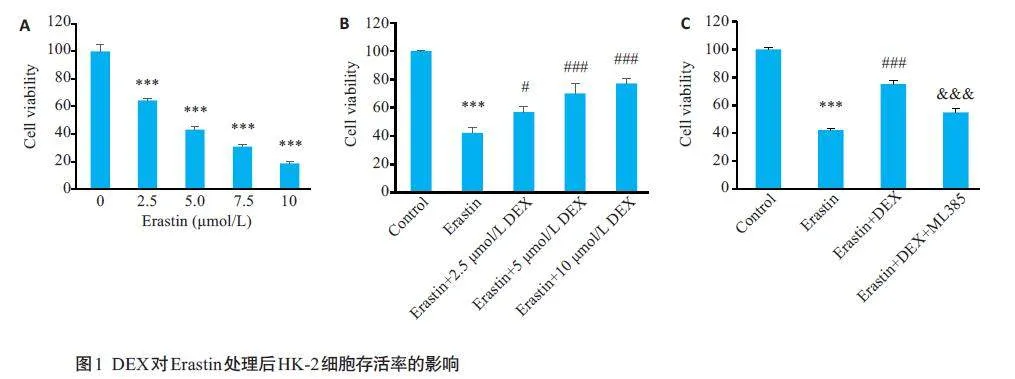
2.1 DEX对Erastin处理后HK-2细胞存活率的影响
随着Erastin 浓度梯度升高,细胞存活率呈现梯度降低(Plt;0.001),当Erastin 浓度为5 μmol/L时,细胞存活率降至50%左右(图1A),因此选取Erastin最佳造模浓度为5 μmol/L。为探究DEX对Erastin诱导HK-2细胞铁死亡的抑制作用,不同浓度DEX(2.5、5、10 μmol/L)预处理2 h后加入5 μmol/L Erastin,数据表明,低、中、高浓度的DEX处理后HK-2细胞存活率相较于Erastin组均有不同程度的升高,且10 μmol/L DEX组的HK-2细胞存活率升高最为明显(Plt;0.001),所以选择10 μmol/L的DEX用于后续实验(图1B)。在10 μmol/L DEX的基础上使用Nrf2 抑制剂ML385,结果发现,ML385 使HK-2 细胞存活率较10 μmol/L DEX组降低(Plt;0.001,图1C),逆转了DEX对HK-2细胞的保护作用。
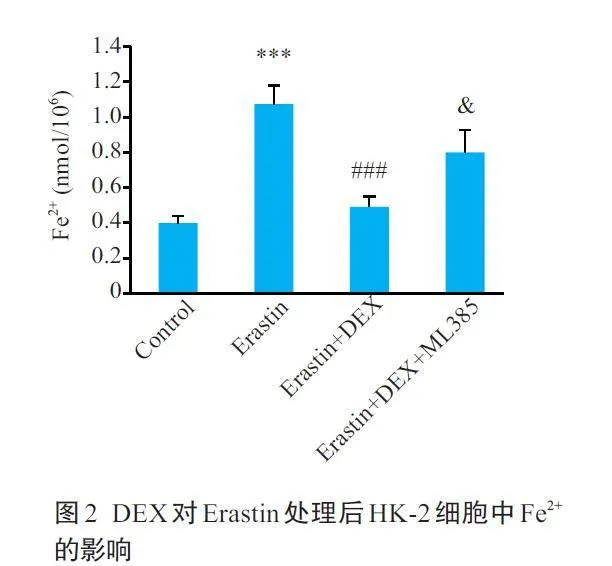
2.2 DEX对Erastin处理后HK-2细胞中Fe2+的影响
与对照组相较,Erastin 组细胞内Fe2+含量增加(Plt;0.001);与Erastin组相较,10 μmol/L DEX使细胞内Fe2+含量明显降低(Plt;0.001);在10 μmol/L DEX的基础上增加Nrf2 抑制剂ML385,数据表明,ML385 使细胞内Fe2+含量较10 μmol/L DEX组升高(Plt;0.05,图2)。
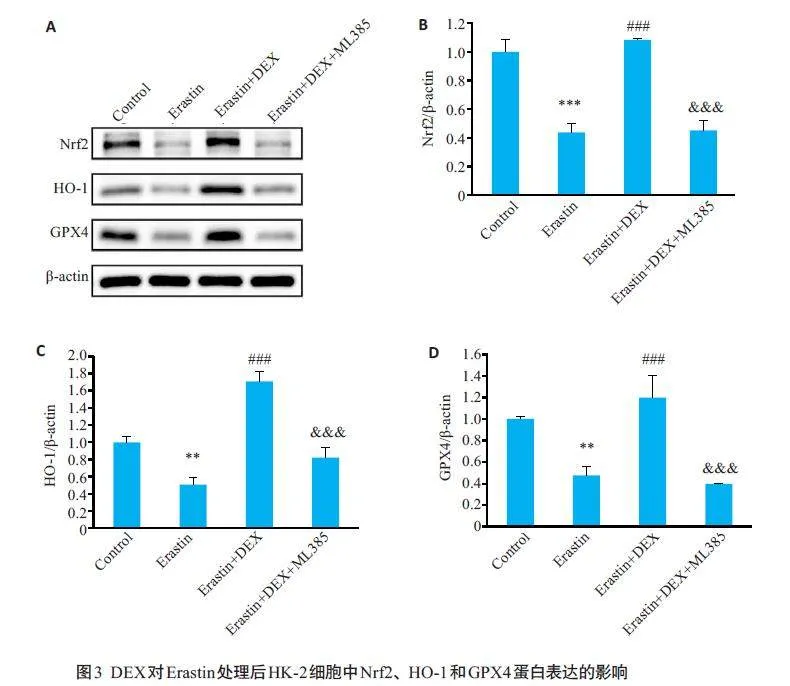
2.3 DEX对Erastin 处理后HK-2 细胞中Nrf2、HO-1 和GPX4蛋白表达的影响
与对照组相较,Erastin组HK-2细胞内Nrf2、HO-1和GPX4 蛋白的表达降低(Plt;0.01);而与Erastin 组相比,10 μmol/L DEX处理组Nrf2、HO-1和GPX4蛋白的表达显著升高(Plt;0.001);而使用Nrf2 抑制剂ML385后,Nrf2/HO-1/GPX4通路被抑制,Nrf2、HO-1和GPX4蛋白的表达降低(Plt;0.001,图3)。
2.4 DEX对Erastin处理后HK-2细胞中ROS的影响
采用流式细胞术检测细胞内ROS 含量,结果显示,与对照组相较,Erastin 组细胞内ROS 的含量升高(Plt;0.001);DEX 可以减弱Erastin 损伤诱导的细胞内ROS 生成(Plt;0.001);而在使用Nrf2 抑制剂ML385后,细胞内ROS 的含量升高(Plt;0.001,图4)。
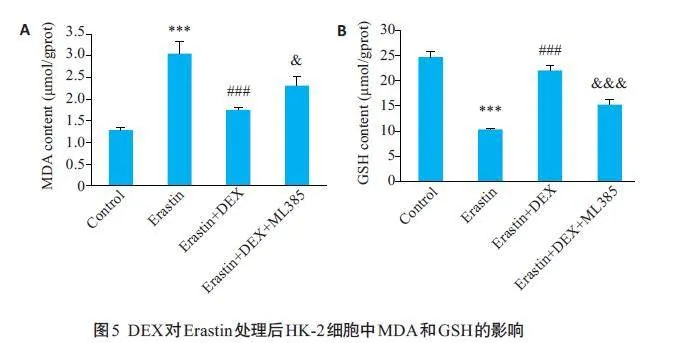
2.5 DEX对Erastin 处理后HK-2 细胞中MDA和GSH的影响
与对照组相较,Erastin组细胞内MDA的含量升高(Plt;0.001,图5A),GSH的含量则降低(Plt;0.001,图5B);与Erastin 组相比,DEX使细胞内MDA的含量明显降低(Plt;0.001,图5A),GSH的含量明显升高(Plt;0.001,图5B);而在DEX 的基础上加用Nrf2 抑制剂ML385后,细胞内MDA的含量升高(Plt;0.05,图5A),GSH的含量则降低(Plt;0.001,图5B)。
3 讨论
近期研究表明,铁死亡与多种肾脏疾病的发生发展密切相关[16-18],因此,早期干预铁死亡的发生,对肾脏病程的截断具有重要的意义。DEX可通过抗氧化应激、抗炎和抑制细胞凋亡等多种途径发挥肾脏保护作用[19, 20]。有文献报道,DEX对肾缺血再灌注损伤的保护作用与其抑制铁死亡有关[21]。目前关于DEX对肾脏疾病的保护作用的研究愈发深入,但DEX对Erastin诱导的HK-2细胞铁死亡是否具有保护作用尚不清楚。为了探究DEX对HK-2 细胞铁死亡的保护作用,我们采用CCK-8法检测不同浓度的DEX对Erastin处理后HK-2细胞存活率的影响。实验结果表明,不同浓度的DEX均能增加Erastin处理后的HK-2细胞存活率,说明DEX能够减轻HK-2细胞铁死亡。加用ML385后,细胞存活率降低,这表明DEX抗HK-2细胞铁死亡的作用可能与其激活Nrf2有关。
当细胞发生铁死亡时,铁稳态的失衡会导致细胞内脂质过氧化的增加[22]。细胞内过量的铁通过芬顿反应产生大量ROS,致使细胞膜中的多不饱和脂肪酸发生脂质过氧化,产生大量脂质过氧化物。其次,铁参与脂氧合酶(LOX)的激活,LOX是含铁酶,可促进脂质过氧化物的产生[23, 24]。细胞内的脂质过氧化导致细胞和线粒体膜损伤和破裂,最终导致铁死亡[25]。另一方面,GSH是细胞内重要的抗氧化剂。GPX4是使用GSH作为还原底物的脂质过氧化的关键调节剂,GPX4可以将脂质过氧化物还原为脂质醇,从而减轻铁死亡[23, 24]。Erastin诱导HK-2 细胞发生铁死亡后,细胞内铁累积、GSH耗竭以及GPX4活性下降,细胞抗氧化能力降低,致使脂质过氧化与代谢功能障碍,ROS和脂质过氧化产物丙二醛(MDA)大量累积。有文献指出,DEX对肾脏的保护作用与其抗氧化的能力有关[26]。本研究发现,DEX可以降低Erastin处理后HK-2细胞内Fe2+的含量,提高细胞内GSH的含量以及GPX4蛋白的表达,使用DEX增强了细胞的抗氧化能力,致使脂质过氧化物的分解增加,细胞内ROS和MDA的含量均下降。
Nrf2的激活在抑制铁死亡的过程中发挥着重要作用,Nrf2是细胞抗氧化反应的关键转录因子,转录调控与铁死亡相关的下游基因HO-1、GPX4 等的表达[27, 28]。有文献报道,通过激活Nrf2 抑制铁死亡不仅可以延缓糖尿病肾病的进展,还能够减轻叶酸或顺铂诱导的急性肾损伤[29-31]。本研究发现,DEX可以提高Erastin处理后HK-2细胞中Nrf2的表达,而Nrf2进一步激活铁死亡相关蛋白HO-1、GPX4 的表达,从而抑制Erastin 诱导的HK-2 细胞发生铁死亡。为了进一步探究Nrf2/HO-1/GPX4 通路是否参与了DEX对HK-2 细胞铁死亡的保护作用,我们加用了Nrf2抑制剂ML385。实验结果表明,Nrf2/HO-1/GPX4 通路的激活被抑制,细胞抗氧化能力降低,细胞内Fe2+和脂质过氧化产物的含量增加,表明Nrf2/HO-1/GPX4通路的抑制降低了DEX对HK-2细胞铁死亡的保护作用。
综上所述,DEX对Erastin 诱导的HK-2 细胞铁死亡具有抑制作用,其机制可能是通过激活Nrf2/HO-1/GPX4通路抑制HK-2细胞发生铁死亡。本研究为DEX减轻HK-2细胞铁死亡的作用提供了一定的实验支撑,为临床上合理使用 DEX 提供了新的理论依据。后续研究将通过体内实验进一步验证DEX通过调控铁死亡治疗肾脏疾病的药效与机制。
参考文献:
[1] Zhang JS, Wang BQ, Yuan SZ, et al. The role of ferroptosis in acutekidney injury[J]. Front Mol Biosci, 2022, 9: 951275.
[2] Thapa K, Singh TG, Kaur A. Targeting ferroptosis in ischemia/reperfusion renal injury[J]. Naunyn Schmiedebergs ArchPharmacol, 2022, 395(11): 1331-41.
[3] Wang Y, Bi R, Quan F, et al. Ferroptosis involves in renal tubular celldeath in diabetic nephropathy[J]. Eur J Pharmacol, 2020, 888:173574.
[4] Zhuo WQ, Wen Y, Luo HJ, et al. Mechanisms of ferroptosis inchronic kidney disease[J]. Front Mol Biosci, 2022, 9: 975582.
[5] Chen X, Li JB, Kang R, et al. Ferroptosis: machinery and regulation[J]. Autophagy, 2021, 17(9): 2054-81.
[6] Rochette L, Dogon G, Rigal E, et al. Lipid peroxidation and ironmetabolism: two corner stones in the homeostasis control offerroptosis[J]. Int J Mol Sci, 2022, 24(1): 449.
[7] Stockwell BR. Ferroptosis turns 10: emerging mechanisms,physiological functions, and therapeutic applications[J]. Cell, 2022,185(14): 2401-21.
[8] Jiang XJ, Stockwell BR, Conrad M. Ferroptosis: mechanisms,biology and role in disease[J]. Nat Rev Mol Cell Biol, 2021, 22(4):266-82.
[9] Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a criticalrole in mitigating lipid peroxidation and ferroptosis[J]. Redox Biol,2019, 23: 101107.
[10]Yang WC, Wang YX, Zhang CG, et al. Maresin1 protect againstferroptosis-induced liver injury through ROS inhibition and Nrf2/HO-1/GPX4 activation[J]. Front Pharmacol, 2022, 13: 865689.
[11] Zhang Y, Liu MM, Zhang YY, et al. Urolithin A alleviates acutekidney injury induced by renal ischemia reperfusion through the p62-Keap1-Nrf2 signaling pathway[J]. Phytother Res, 2022, 36(2):984-95.
[12]Zhao Y, Feng XJ, Li B, et al. Dexmedetomidine protects againstlipopolysaccharide-induced acute kidney injury by enhancingautophagy through inhibition of the PI3K/AKT/mTOR pathway[J].Front Pharmacol, 2020, 11: 128.
[13]Shan XS, Zhang JX, Wei X, et al. Dexmedetomidine attenuates renalischemia-reperfusion injury through activating PI3K/Akt-eNOSsignaling via α2 adrenoreceptors in renal microvascular endothelialcells[J]. FASEB J, 2022, 36(11): e22608.
[14]Song L, Feng SL, Yu H, et al. Dexmedetomidine protects against kidney fibrosis in diabetic mice by targeting miR-101-3p-mediatedEndMT[J]. Dose Response, 2022, 20(1): 15593258221083486.
[15]Wang Z, Wu JL, Hu ZL, et al. Dexmedetomidine alleviateslipopolysaccharide-induced acute kidney injury by inhibitingp75NTR-mediated oxidative stress and apoptosis[J]. Oxid MedCell Longev, 2020, 2020: 5454210.
[16]Feng Q, Yu XY, Qiao YJ, et al. Ferroptosis and acute kidney injury(AKI): molecular mechanisms and therapeutic potentials[J]. FrontPharmacol, 2022, 13: 858676.
[17]Mengstie MA, Seid MA, Gebeyehu NA, et al. Ferroptosis in diabeticnephropathy: mechanisms and therapeutic implications[J]. MetabolOpen, 2023, 18: 100243.
[18]Zhou YJ, Zhang JL, Guan QY, et al. The role of ferroptosis in thedevelopment of acute and chronic kidney diseases[J]. J CellPhysiol, 2022, 237(12): 4412-27.
[19]Zhai MY, Han MM, Huang X, et al. Dexmedetomidine protectshuman renal tubular epithelial HK-2 cells against hypoxia/reoxygenation injury by inactivating endoplasmic reticulum stresspathway[J]. Cell J, 2021, 23(4): 457-64.
[20]Liu YT, Liu W, Wan ZH, et al. Protective effect of dexmedetomidineagainst renal injury in diabetic nephropathy rats through inhibitingNF‑κB pathway[J]. Eur Rev Med Pharmacol Sci, 2020, 24(22):11865-70.
[21]Tao WH, Shan XS, Zhang JX, et al. Dexmedetomidine attenuatesferroptosis-mediated renal ischemia/reperfusion injury andinflammation by inhibiting ACSL4 via α2-AR[J]. Front Pharmacol,2022, 13: 782466.
[22]Chen X, Comish PB, Tang DL, et al. Characteristics and biomarkersof ferroptosis[J]. Front Cell Dev Biol, 2021, 9: 637162.
[23]Xie LH, Fefelova N, Pamarthi SH, et al. Molecular mechanisms offerroptosis and relevance to cardiovascular disease[J]. Cells, 2022,11(17): 2726.
[24]Liu J, Kang R, Tang DL. Signaling pathways and defensemechanisms of ferroptosis[J]. FEBS J, 2022, 289(22): 7038-50.
[25]Yan B, Ai YW, Sun Q, et al. Membrane damage during ferroptosis iscaused by oxidation of phospholipids catalyzed by theoxidoreductases POR and CYB5R1[J]. Mol Cell, 2021, 81(2): 355-69. e10.
[26]Chen YP, Feng XJ, Hu XY, et al. Dexmedetomidine amelioratesacute stress-induced kidney injury by attenuating oxidative stressand apoptosis through inhibition of the ROS/JNK signaling pathway[J]. Oxid Med Cell Longev, 2018, 2018: 4035310.
[27]Adedoyin O, Boddu R, Traylor A, et al. Heme oxygenase-1 mitigatesferroptosis in renal proximal tubule cells[J]. Am J Physiol RenalPhysiol, 2018, 314(5): F702-14.
[28]黄庆洋, 纪东东, 田绣云, 等. 小檗碱通过激活Nrf2-HO-1/GPX4 通路抑制小鼠海马神经元HT22 细胞的铁死亡[J]. 南方医科大学学报, 2022, 42(6): 937-43.
[29]Li SW, Zheng LS, Zhang J, et al. Inhibition of ferroptosis by upregulatingNrf2 delayed the progression of diabetic nephropathy[J].Free Radic Biol Med, 2021, 162: 435-49.
[30]Li X, Zou Y, Xing J, et al. Pretreatment with roxadustat (FG-4592)attenuates folic acid-induced kidney injury through antiferroptosisvia akt/GSK-3β/Nrf2 pathway[J]. Oxid Med Cell Longev, 2020,2020: 6286984.
[31]Meng XY, Huang WJ, Mo WW, et al. ADAMTS-13-regulatednuclear factor E2-related factor 2 signaling inhibits ferroptosis toameliorate cisplatin-induced acute kidney injuy[J]. Bioengineered,2021, 12(2): 11610-21.
(编辑:经 媛)
基金项目:安徽省自然科学研究重点项目基金(KJ2021A0785);蚌埠医学院研究生科研创新计划项目基金支持(Byycx22100)
猜你喜欢
现代临床医学(2021年6期)2021-11-20 06:34:50
建材发展导向(2021年11期)2021-07-28 06:57:22
当代水产(2020年10期)2020-03-17 07:02:48
当代水产(2019年8期)2019-10-12 08:57:26
中成药(2018年9期)2018-10-09 07:18:36
中成药(2018年1期)2018-02-02 07:19:53
中成药(2017年12期)2018-01-19 02:06:48
中成药(2017年4期)2017-05-17 06:09:26
中国洗涤用品工业(2015年4期)2015-02-28 19:02:15
中成药(2014年9期)2014-02-28 22:28:50
