
一测多评法测定甘草及其不同炮制品中7种成分含量
2024-10-25 00:00冯玉郗仲玟麻景梅高乐牛丽颖田宇柔
河北工业科技 2024年5期

摘 要:为了实现对甘草药材及其炮制品成分的多指标质量评价,建立了一测多评测定甘草药材及3种炮制品中7种化学成分含量的方法。以甘草苷为内标,计算异甘草苷相对校正因子;以甘草酸为内标,计算芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的相对校正因子,再测定各成分含量,并将计算结果与外标法测定结果进行比较。结果表明:一测多评法测定甘草及其不同炮制品中7种成分的含量具有可行性;甘草炮制后,其中的甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷的含量均有所降低,且在不同炮制品中的含量呈现出相同的变化趋势,均为ω(甘草药材)>ω(炒甘草)>ω(甘草片)>ω(炙甘草)。所建立的一测多评法稳定、准确,且重复性好,可为甘草及其不同炮制品的质量评价提供参考。
关键词:中药化学;甘草;炮制品;一测多评;质量评价
中图分类号:R284.1 文献标识码:A
Determination of seven components in licorice and its different
processed products by QAMS
Abstract:
In order to achieve the quality evaluation of multiple indicator components in licorice and its processed products, a quantitative analysis of multi-components by single marker (QAMS) was established to determine the content of seven chemical components in licorice medicinal materials and three different processed products. The calibration factor for isoliquiritigenin was calculated by using glycyrrhizin as the internal standard; The relative correction factors of apigenin glycyrrhizic acid, apigenin isoliquiritigenin, neoisoliquiritigenin, and glycyrrhizin were calculated by using glycyrrhetinic acid as the internal standard, and then the content of each component was determined. The calculated results were compared with the results obtained by the external standard method. The results show that the QAMS is feasible for determining the content of seven components in licorice and its different processed products. After processing, the content of glycyrrhizin, glycyrrhetinic acid, apigenin glycyrrhizin, apigenitigenin, and neoisoglycyrrhizin all decreases and shows the same trend of change. The trend of content change is ω (licorice herbs)> ω (fried licorice)> ω (licorice tablets)> ω (roasted licorice). The established method is stable, accurate, and has good repeatability, which can provide reference for the quality evaluation of licorice and its different processed products.
Keywords:
chemistry of Chinese materia medica; licorice; processed products; QAMS; quality evaluation
甘草为豆科植物甘草(Glycyrrhiza uralensis Fisch.)、胀果甘草(Glycyrrhiza inflata Bat.)或光果甘草(Glycyrrhiza glabra L.)的干燥根及根茎,具有补脾益气、清热解毒、祛痰止咳、缓急止痛、调和诸药之功效[1]。甘草临床应用广泛,素有“十方九草,无草不成方”之说。甘草炮制方法较多,经历代发展,现代市售甘草炮制品主要有3种,即甘草片、炙甘草和炒甘草。甘草片和炙甘草为《中华人民共和国药典》[1](以下简称《中国药典》)收载的甘草炮制品,炒甘草多收载于省级炮制规范[2-5]。甘草片味甘性偏凉,长于泻火解毒、化痰止咳;炙甘草味甘性温,长于补脾益气;炒甘草味甘性燥,长于补中益气、养正和中。
对于甘草不同炮制品的研究虽有报道,但多是采用HPLC指纹图谱或多成分定量的方法对甘草炮制前后化学成分的含量进行比较研究。例如:陈佳等[6]采用化学模式,识别分析了甘草药材及炙甘草炮制前后化学成分的差异;宁雪等[7]建立了同时测定甘草及其不同炮制品中甘草苷、异甘草苷、甘草素等7种成分含量的HPLC法。一测多评法(quantitative analysis of multi-components by single-marker,QAMS)仅用一个对照品就能够实现多指标成分的测定。因此,该法已被广泛应用于中药及其制剂的成分含量测定中[8],比如:裴玉琼等[9]采用QAMS法测定了炙甘草饮片中6种成分的含量。但是,现有文献报道中尚未见采用一测多评法对甘草药材及3种市售炮制品中有效成分含量进行研究的报道。因此,本研究采用一测多评法测定甘草药材及其3种炮制品(甘草片、炙甘草、炒甘草)中甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的含量,分析这些成分在甘草及其炮制品中的含量变化,为甘草及其炮制品的质量控制提供参考。
1 材 料
1.1 仪 器
高效液相色谱仪:LC-15C,岛津公司提供;Waters e2695,沃特世科技(上海)有限公司提供;Agilent 1260Ⅱ,安捷伦科技有限公司提供。
色谱柱:Symmetry C18(4.6 mm×250 mm,5 μm),沃特世科技(上海)有限公司提供;Wondasil C18(4.6 mm×250 mm,5 μm),岛津公司提供;ZORBAX Eclipse XDB-C18(4.6 mm×250 mm,5 μm),安捷伦科技有限公司提供。
电子天平(BSA224S-CW,CPA225D),赛多利斯公司提供;KQ-250E超声波清洗器(250 W,40 kHz),昆山市超声仪器有限公司提供。
1.2 试 药
甘草素(批号为DST171014-010,纯度≥98%),成都德思特生物技术有限公司提供;甘草酸铵(批号为110731-201619,纯度≥93.0%),甘草苷(批号为11610-201607,纯度≥93.1%),均由中国食品药品检定研究院提供;异甘草苷(批号为18101805,纯度≥98%),新异甘草苷(批号为18102405,纯度≥98%),芹糖异甘草苷(批号为18102407,纯度≥98fgfXCtc1cYesFUo5qNDFuD9kvayaDW7m2TMpM0RF7SI=%),芹糖甘草苷(批号为18102403,纯度≥98%),均由成都普菲德生物技术有限公司提供。
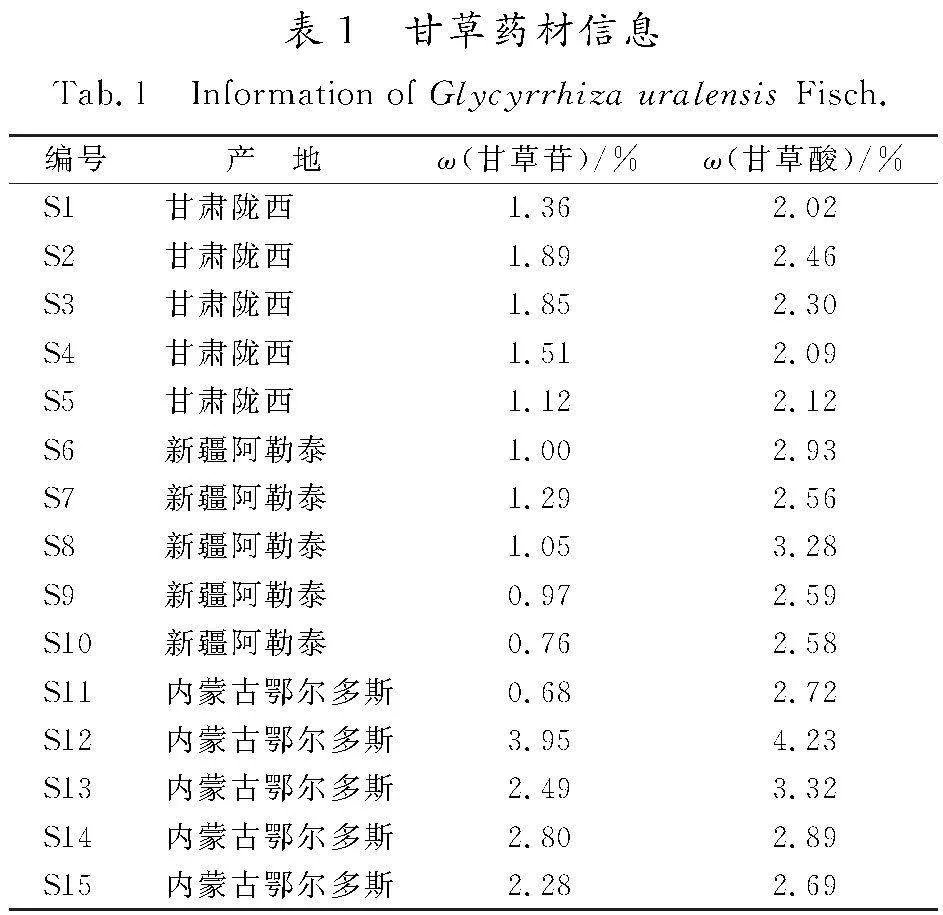
15批甘草药材(编号为S1—S15)经神威药业集团有限公司质量检测中心鉴定为豆科植物甘草(Glycyrrhiza uralensis Fisch.)的干燥根及根茎,按照《中国药典》进行检测,结果均符合要求,药材具体信息见表1。甘草炮制品为本实验室自行制备。
2 方法和结果
2.1 前期准备
2.1.1 炮制品的制备
1)甘草片
参照《中国药典》“甘草”项下甘草片炮制方法,从15批甘草药材中各取约200 g,除去杂质,洗净,润透,切厚片,干燥,即得(编号为P1—P15)。
2)炙甘草
参照《中国药典》“甘草”项下炙甘草炮制方法,取按照本节“1)”项下制备方法所制甘草片,15批各约200 g,照蜜炙法炒至黄色至深黄色,不粘手时取出,晾凉(编号为Z1—Z15)。
3)炒甘草
参照《山东省中药饮片炮制规范》[4]“炒甘草”项下炮制方法,取按照本节“1)”项下
制备方法所制甘草片,15批各约200 g,置于已预热的炒制容器内,文火加热,炒至表面深黄色,取出,晾凉(编号为C1—C15)。
2.1.2 溶液的制备
1)对照品溶液的制备
分别取芹糖甘草苷、甘草苷、芹糖异甘草苷、异甘草苷、新异甘草苷、甘草素、甘草酸对照品适量,精密称定,加70%(体积分数,下同)乙醇
制成每1 mL分别含芹糖甘草苷0.053 0 mg、甘草苷0.025 7 mg,芹糖异甘草苷0.062 2 mg,异甘草苷0.051 7 mg,新异甘草苷0.050 9 mg,甘草素0.014 8 mg,甘草酸0.203 9 mg的混合对照品溶液。
2)供试品溶液的制备
分别取甘草药材、甘草片、炙甘草、炒甘草饮片粉碎后过4号筛,各取约0.2 g,精密称定,置于50 mL具塞锥形瓶中,精密加入70%乙醇25 mL,再称定质量,超声处理30 min,放冷,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液,0.22 μm微孔滤膜过滤,即得。
2.1.3 色谱条件的确定
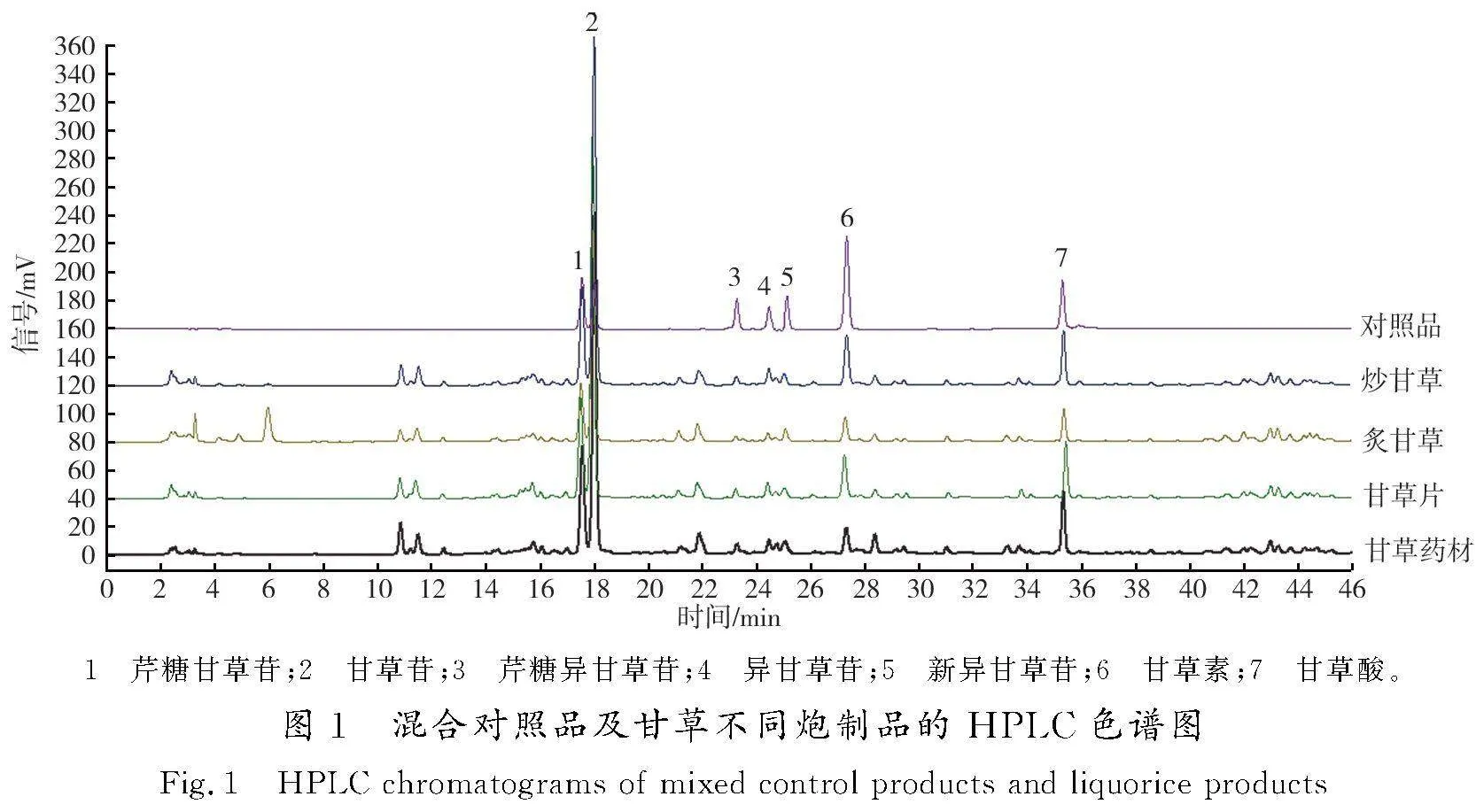
测定时选用Symmetry C18色谱柱(4.6 mm×250 mm,5 μm);柱温为35 ℃;流动相为乙腈(A)-0.1%磷酸(B)。其梯度洗脱程序:0~<20 min,10%~<30%A;20~<45 min,30%~<65%A;45~65 min,65%~95%A。流速为1.0 mL/min;检测波长为276 nm。理论板数按甘草苷峰计算,应不低于5 000。混合对照品及甘草不同炮制品的液相色谱图见图1。
2.2 方法学考察
2.2.1 线性范围考察
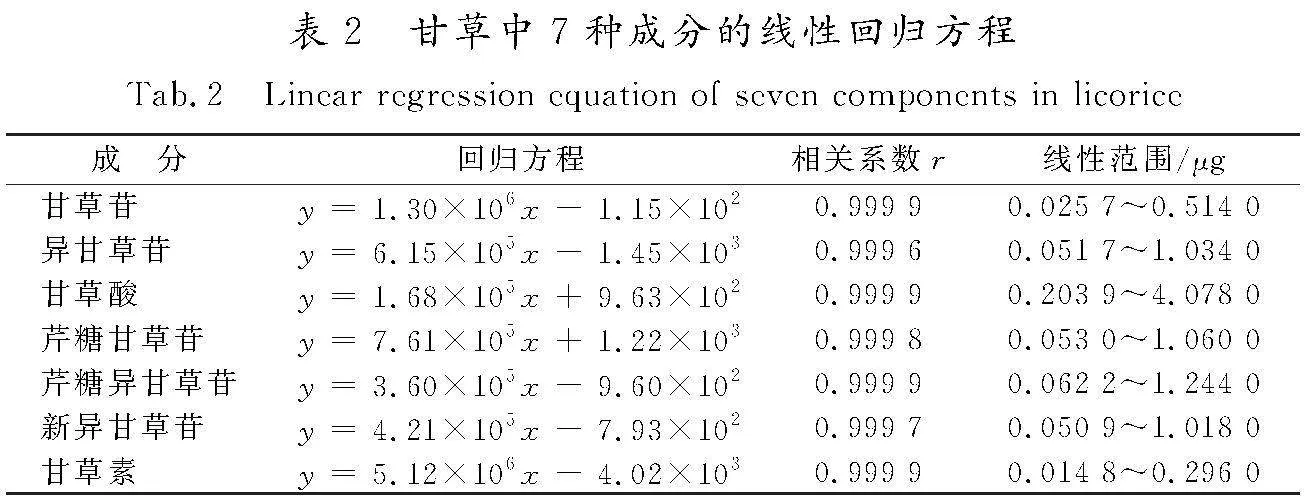
精密吸取混合对照品溶液1,4,8,10,14,16,20 μL,分别注入到高效液相色谱仪,以进样量作为横坐标(X),峰面积作为纵坐标(Y),进行线性回归,绘制标准曲线,得到甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的线性回归方程,如表2所示。
2.2.2 精密度试验
精密吸取按照“2.1.2”项下制备方法制得的混合对照品溶液,按“2.1.3”项下色谱条件连续进样测定6次,记录甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的峰面积,计算各成分峰面积的RSD值分别为0.35%,1.04%,0.76%,0.49%,1.01%,0.90%,1.08%,表明仪器的精密度良好。
2.2.3 稳定性试验
取甘草药材(编号为S1)粉末约0.2 g,精密称定,按“2.1.2”项下制备方法制备供试品溶液,分别于0,4,8,12,18,24 h进样,每次进样10 μL,记录甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的峰面积,计算各成分峰面积的RSD值分别为1.21%,0.70%,1.12%,1.19%,0.86%,0.93%,1.10%,表明供试品溶液在24 h内稳定性良好。
2.2.4 重复性试验
取同一批样品(编号为S1)约0.2 g,精密称定,按“2.1.2”项下方法平行制备6份供试品溶液,按“2.1.3”项下色谱条件进样测定,记录峰面积,计算甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素含量的RSD值,结果分别为1.23%,0.98%,0.90%,1.00%,1.14%,1.07%,1.23%,表明该方法的重复性良好。
2.2.5 加样回收率试验
取已知含量的样品粉末9份(编号为S1),每份约0.1 g,精密称定,置于50 mL具塞锥形瓶中,分别按各成分含量(质量分数,下同)的80%,100%,120%精密加入甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的对照品储备液,按“2.1.2”项下方法制备供试品溶液,按 “2.1.3”项下色谱条件进样测定,计算加样回收率。各成分的平均加样回收率(括号外数值)和RSD值(括号内数值)分别为102.06%(1.28%)、101.51%(2.40%)、101.72%(2.02%)、100.65%(2.72%)、101.02%(2.72%)、102.68%(1.87%)、101.61%(2.14%)。
2.3 相对校正因子评价
2.3.1 相对校正因子的计算
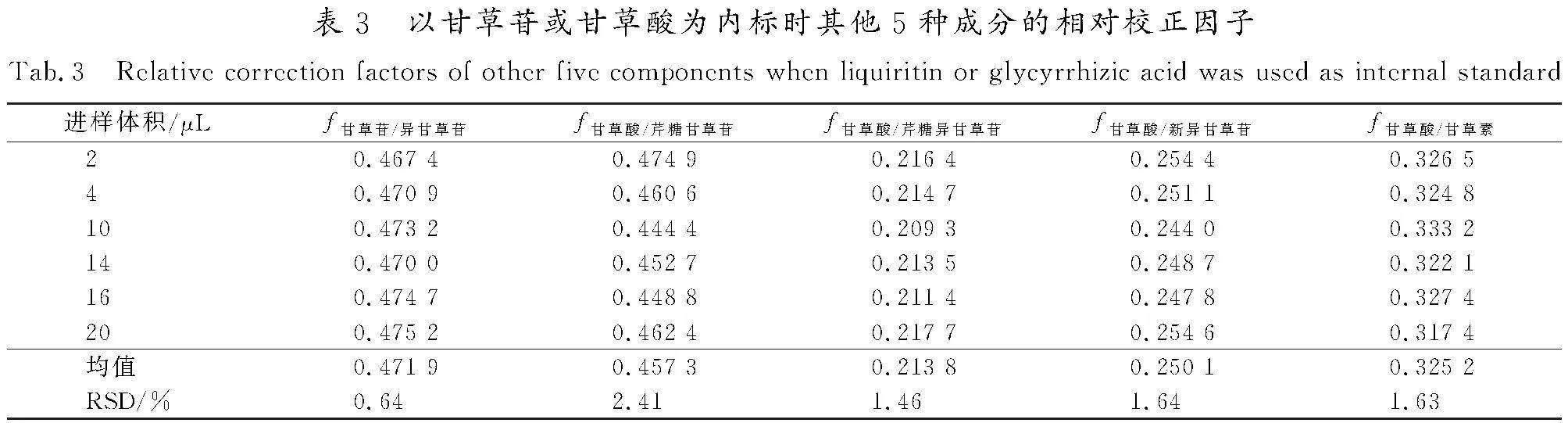
分别取2,4,10,14,16,20 μL混合对照品溶液进样,并将检测结果按公式fk/m=fk/fm=(Wk×Am)/(Wm×Ak)进行计算,得到相对校正因子。式中:Ak为内标物的峰面积;Wk为内标物的质量;Am为其他组分m的峰面积;Wm为其他组分的质量[10-13]。以甘草苷为内标,计算异甘草苷的校正因子;以甘草酸为内标,分别计算芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的相对校正因子,结果见表3。
2.3.2 高效液相色谱仪及色谱柱的考察
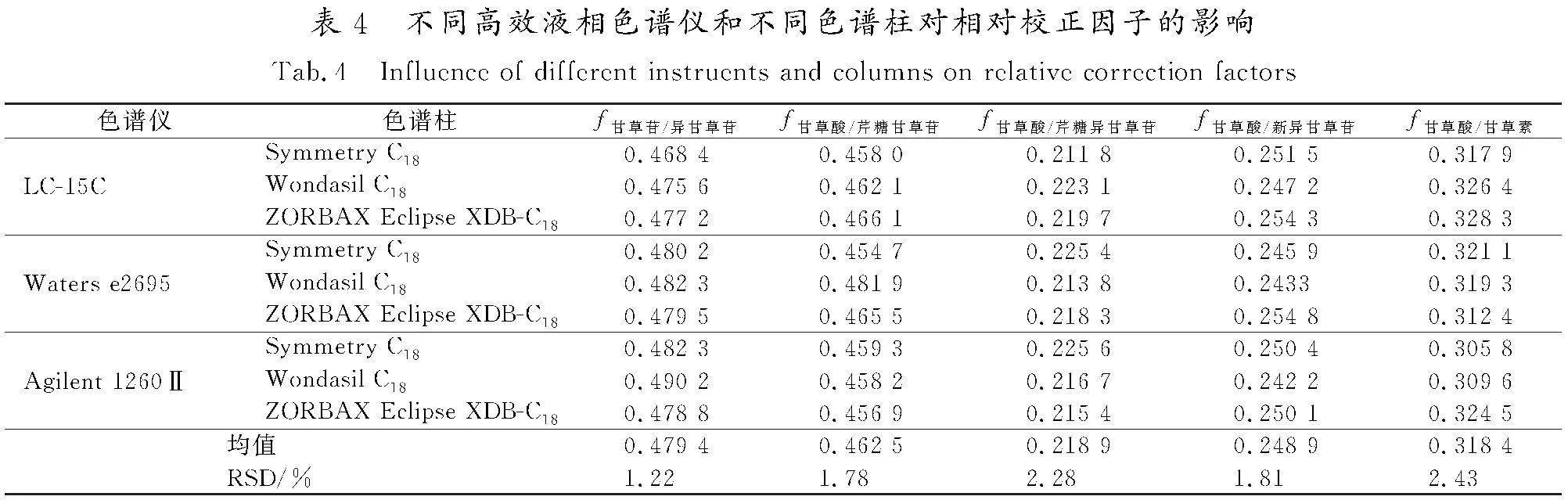
分别使用不同品牌的高效液相色谱系统(LC-15C,Waters e2695和Agilent 1260Ⅱ),以及Symmetry C18(4.6 mm×250 mm,5 μm)、Wondasil C18(4.6 mm×250 mm,5 μm)和ZORBAX Eclipse XDB-C18(4.6 mm×250 mm,5 μm)3种不同的色谱柱,考察高效液相色谱仪及色谱柱对相对校正因子的影响。结果显示,不同仪器、不同色谱柱对相对校正因子无显著性影响(结果见表4),以耐用性结果最终确定f甘草苷/异甘草苷=0.48,f甘草酸/芹糖甘草苷=0.46,f甘草酸/芹糖异甘草苷=0.22,f甘草酸/新异甘草苷=0.25,f甘草酸/甘草素=0.32。
2.3.3 待测组分色谱峰的定位
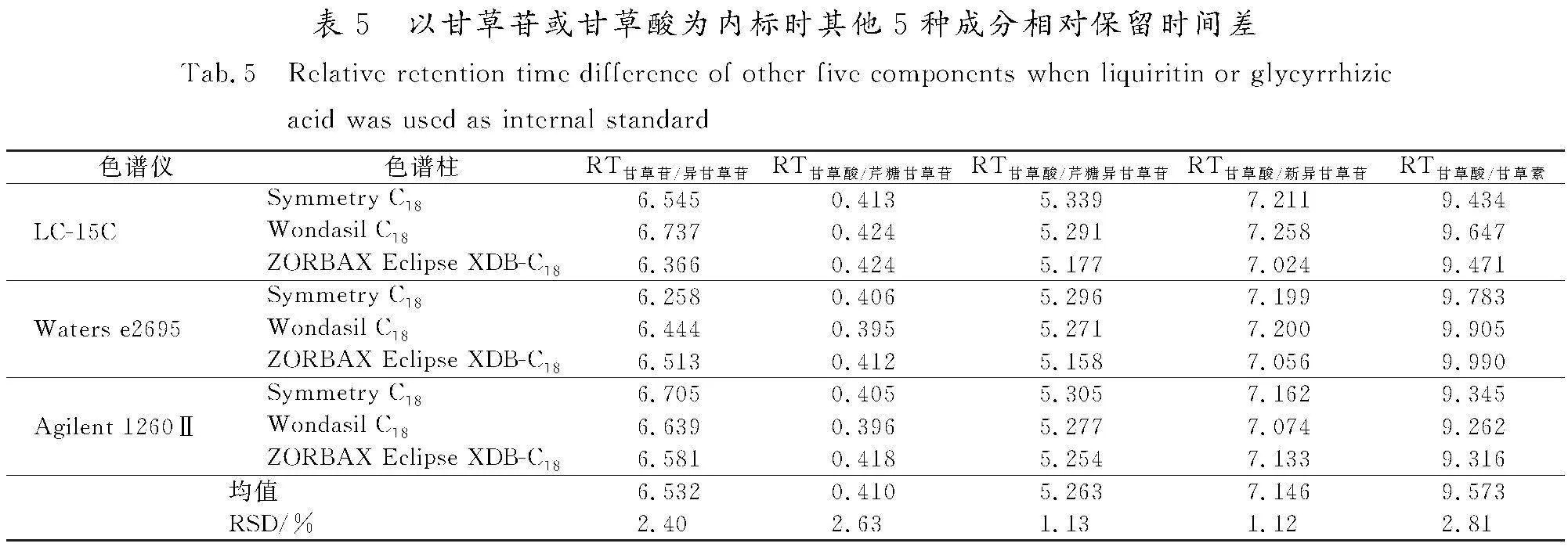
采用不同高效液相色谱仪及色谱柱时,待测组分的保留时间会有所波动,待测组分的色谱峰,一般根据相对保留时间差(RT内标物/待测=RT待测-RT内标物)进行定位[11]。取按照“2.1.2”项下制备方法制得的混合对照品溶液,分别采用3种色谱仪与不同色谱柱测定,以甘草苷为内标,计算异甘草苷的相对保留时间差;以甘草酸为内标,分别计算芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的相对保留时间差,结果(见表5)显示5种成分相对保留时间差的RSD值均小于3%,表明利用相对保留时间差可定位色谱峰。
2.4 一测多评法与外标法的结果比较
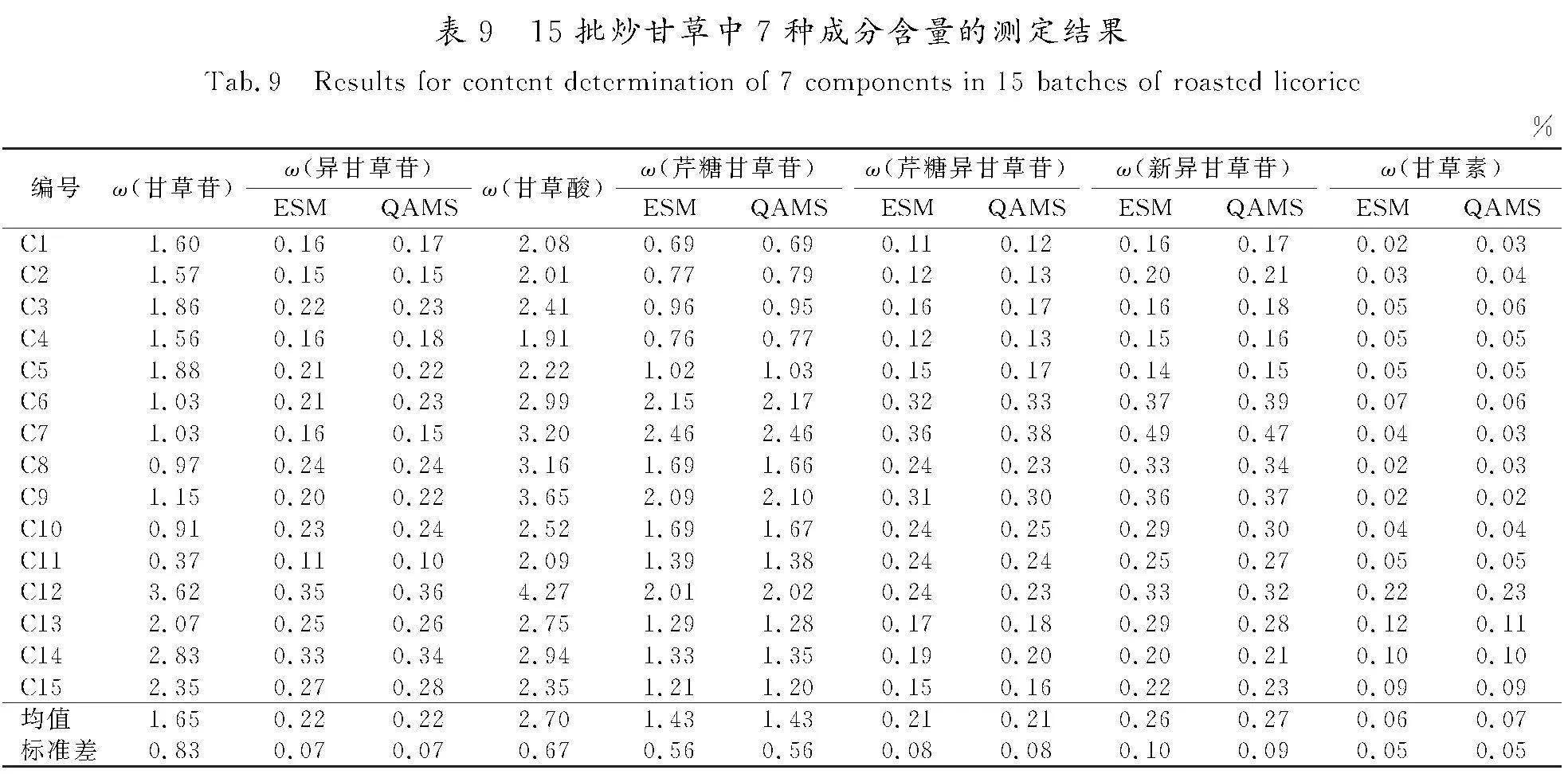
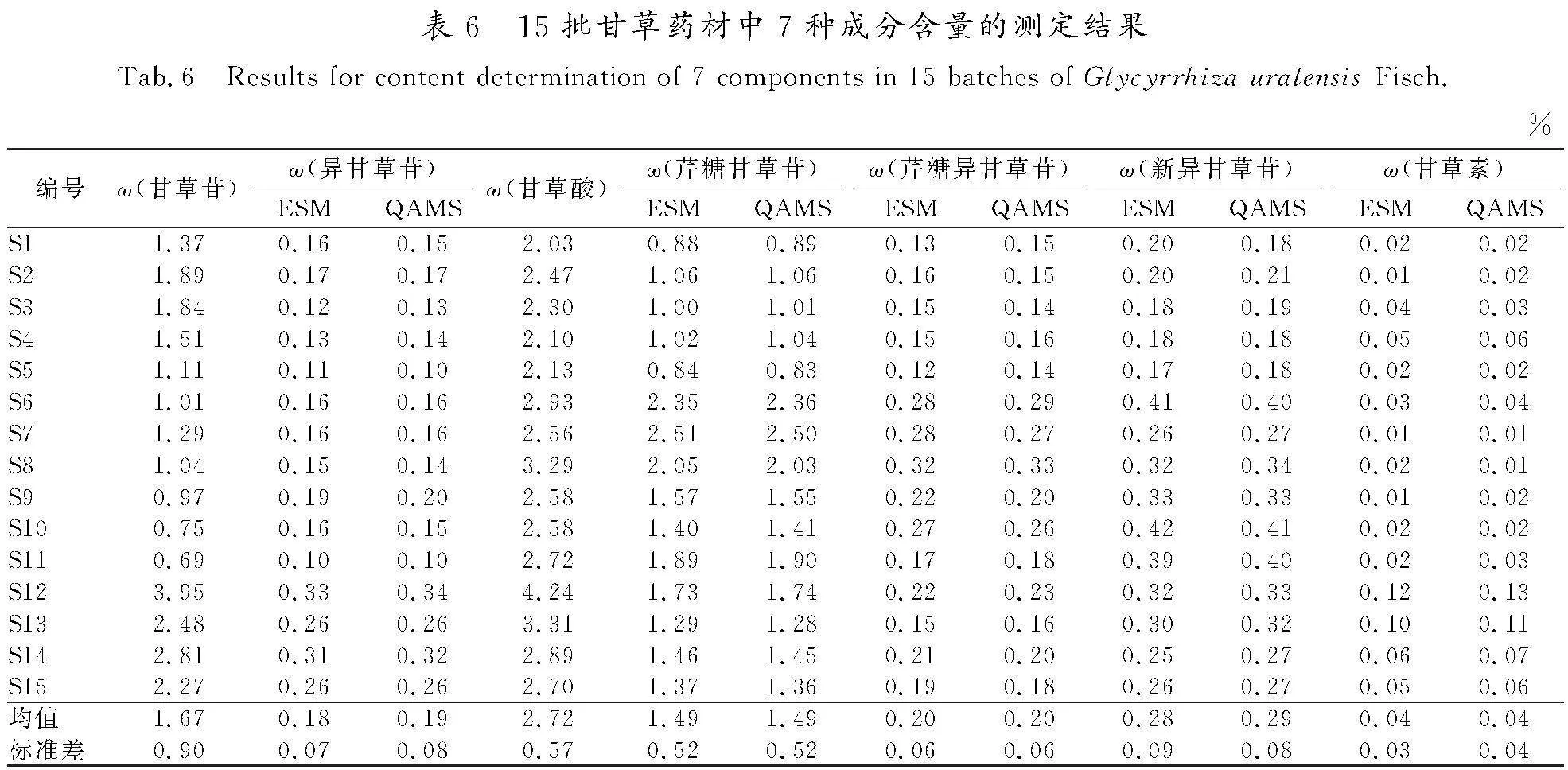
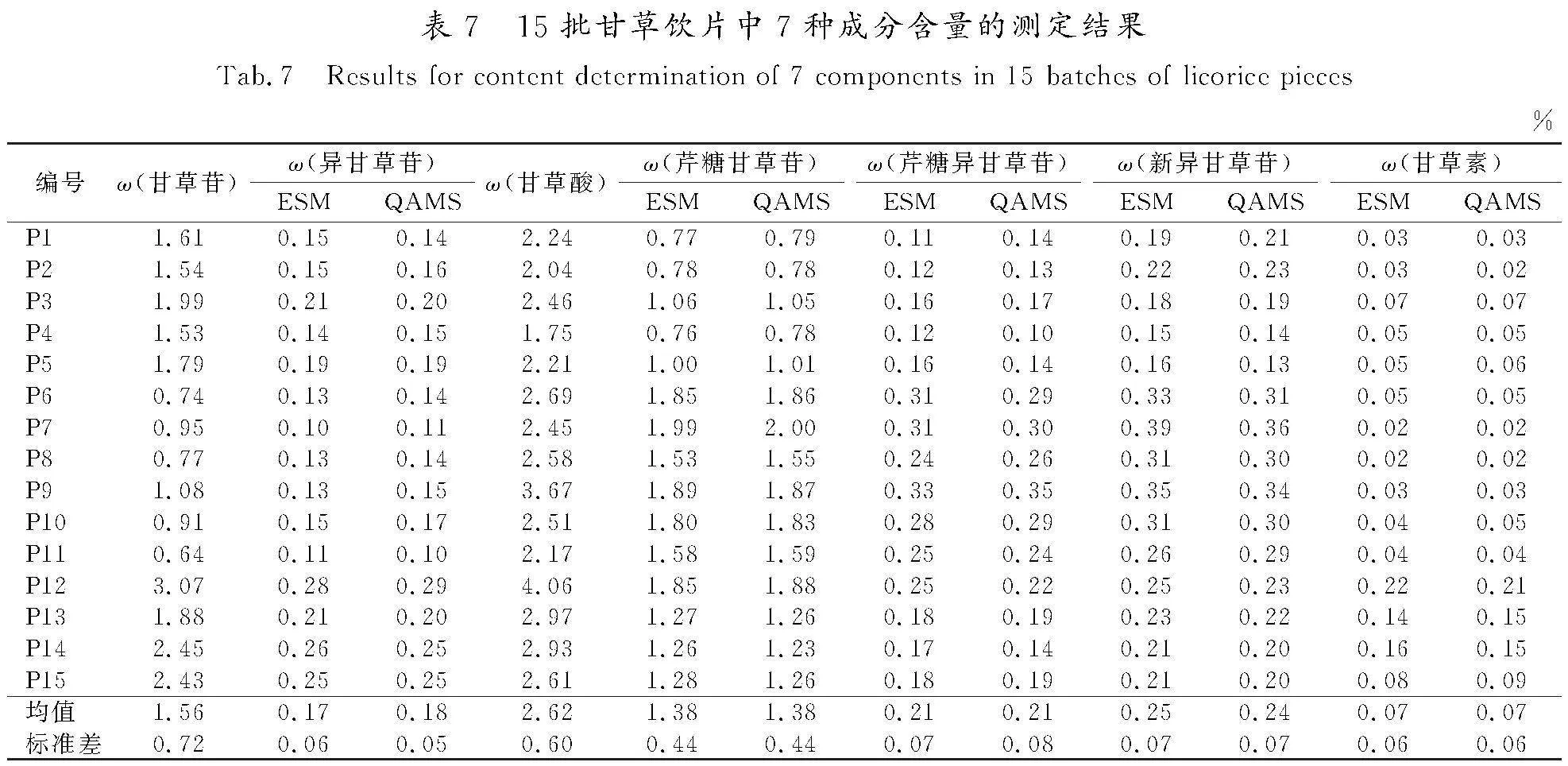
分别取15批甘草药材按照“2.1.2”项下方法制备供试品溶液,按“2.1.3”项下色谱条件测定。分别采用外标法(external standard method,ESM)与一测多评法计算甘草药材中甘草苷、异甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷、甘草素的含量。结果(见表6—表9)表明,2种方法测得的各成分含量无明显差异,甘草药材中的甘草苷、甘草酸含量与采用《中国药典》方法所得检测结果(详见表1)无明显差异。这说明将QAMS应用于甘草药材及其炮制品的多成分指标质量评价中是可行的。甘草经炮制后,其成分中的甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷的含量均有所降低,且在不同炮制品中呈现出相同的变化趋势,均为ω(甘草药材)>ω(炒甘草)>ω(甘草片)>ω(炙甘草),此变化趋势与文献报道中关于甘草药材及其炮制品的含量变化趋势是一致的[14-15];甘草素含量的变化趋势为ω(甘草片)>ω(炙甘草)>ω(炒甘草)>ω(甘草药材),异甘草苷含量的变化趋势为ω(炒甘草)>ω(甘草药材)>ω(甘草片)>ω(炙甘草)。
2.5 讨 论
本研究对选用的3个产地15批甘草药材按照《中国药典》方法进行了检测,结果均符合要求。在实验前期,分别对流动相、提取溶剂和超声提取时间进行了优选。首先,对乙腈-0.1%甲酸、乙腈-0.1%磷酸、甲醇-0.1%磷酸等3个流动相系统进行对比研究,发现以乙腈-0.1%磷酸为流动相时,各色谱峰分离度良好、基线平稳、保留时间适中。故本研究选取乙腈-0.1%磷酸为流动相进行梯度洗脱。其次,将甘草药材粉末过40目(0.425 mm)筛对供试品溶液制备方法进行了考察,分别选用50%甲醇、70%甲醇、甲醇(分析纯)、稀乙醇、70%乙醇和乙醇(分析纯)为提取溶剂,分别以10,30,45 min为超声提取时间进行对比研究。结果显示,采用70%乙醇作为提取溶剂并超声提取30 min时,各成分含量最高。随后,将此提取条件与加热回流提取方式进行了对比,结果并无明显差异。因此,确定供试品溶液制备方法为以70%乙醇为提取溶剂,并超声提取30 min。
因中药具有多成分、多功效的特点,近年来多采用多成分同时定量的模式进行质量控制[16-17],但高昂的检测成本又限制了多指标质量控制模式的应用。一测多评法是采用相对易得、价廉的内参对照品,实现对多成分同时定量的分析方法[11-12]。本研究建立了一测多评法测定甘草药材及其3种炮制品中7种化学成分的含量测定方法。方法学考察结果表明,所建立方法的重复性和稳定性的RSD值均小于3%,平均加样回收率为95%~105%;各待测成分与内标物的相对校正因子和相对保留时间在不同品牌高效液相色谱仪和不同色谱柱上得到了较好的验证,RSD值均小于3%,说明方法的重现性良好。通过对15批次甘草及其不同炮制品的检测,一测多评法与外标法的含量测定结果无显著差异,并且测定结果显示甘草经炮制后的成分中,甘草苷、甘草酸、芹糖甘草苷、芹糖异甘草苷、新异甘草苷的含量均有所降低,且在不同炮制品中呈现出相同的变化趋势,均为ω(甘草药材)>ω(炒甘草)>ω(甘草片)>ω(炙甘草)。
3 结 语
本研究采用一测多评法测定了甘草及其不同炮制品中7种化学成分的含量,分别对流动相、检测波长、色谱柱、提取溶媒、提取方法、提取时间等进行了考察,最终确定了甘草及其不同炮制品中多成分含量测定的最优条件。方法学结果表明,甘草苷、异甘草苷、甘草酸等7种成分在一定范围内线性关系良好,r值均达到0.999 6及以上,回收率(n=9)均为95%~105%。所建立的一测多评法具有准确性高、供试品制备方法简单、节约成本等优点,可为甘草及其不同炮制品的质量评价提供可靠依据。
由甘草药材及其不同炮制品的含量测定结果可知,甘草炮制前后各化学成分的变化趋势不同,表明甘草在炮制过程中化学成分的变化是复杂的,但仅对7种化学成分含量进行研究尚不能充分、科学地阐述甘草炮制的科学内涵,后期应全面比较不同炮制方法对甘草化学成分的影响,探讨甘草不同炮制品物质基础的差异性,为阐明甘草炮制的科学内涵及更合理的临床应用提供参考。
参考文献/References:
[1] 国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2010:88-89.
[2] 上海市卫生局.上海市中药饮片炮制规范[M].上海:上海科学技术出版社,1983:41.
[3] 广西壮族自治区食品药品监督管理局.广西壮族自治区中药饮片炮制规范[M].南宁: 广西科学技术出版社,2022:41.
[4] 山东省食品药品监督管理局,山东省食品药品检验研究院.山东省中药饮片炮制规范[M].济南: 山东科学技术出版社,2022:83-84.
[5] 浙江省食品药品监督管理局.浙江省中药炮制规范[M].北京: 中国医药科技出版社, 2015: 23.
[6] 陈佳,张权,杨蕊,等.甘草药材及其炮制品炙甘草化学模式识别分析[J].药物分析杂志,2020,40(11):1963-1976.
CHEN Jia,ZHANG Quan,YANG Rui,et al.Chemical pattern recognition of Glycyrrhiza uralensis Fisch. and its processed products Glycyrrhizae Radix et Rhizoma Praeparata cum melle[J].Chinese Journal of Pharmaceutical Analysis,2020,40(11):1963-1976.
[7] 宁雪,杨振泳,王凤文,等.HPLC同时测定甘草及其炮制品中7种成分的含量[J].中药材,2020,43(9):2146-2150.
[8] 何华康,刘芳,杨彬,等.基于CNKI数据库的"一测多评"相关文献计量学分析[J].海峡药学,2019,31(10):48-52.
HE Huakang,LIU Fang,YANG Bin,et al.Bibliometrics analysis of quantitative analysis of multi-components by single-maker based on CNKI database[J].Strait Pharmaceutical Journal,2019,31(10):48-52.
[9] 裴玉琼,石海培,严辉,等.基于HPLC指纹图谱及多成分一测多评法定量的炙甘草饮片质量评价研究[J].中草药,2019,50(18):4293-4304.
PEI Yuqiong,SHI Haipei,YAN Hui,et al.Quality evaluation of Glycyrrhizae Radix et Rhizoma Praeparata Cum Melle based on HPLC fingerprint and multi-component quantitative analysis by QAMS[J].Chinese Traditional and Herbal Drugs,2019,50(18):4293-4304.
[10]王智民,钱忠直,张启伟,等.一测多评法建立的技术指南[J].中国中药杂志,2011,36(6):656-658.
[11]孟俊华,刘媛,占慧慧,等.基于特征图谱结合化学计量学和一测多评法的薤白质量评价[J].中草药,2023,54(21):7176-7185.
MENG Junhua,LIU Yuan,ZHAN Huihui,et al.Quality evaluation of Allii Macrostemonis Bulbus based on fingerprint HPLC spectrum combined with chemometrics and quantitative analysis of multi-components with a single-marker[J].Chinese Traditional and Herbal Drugs,2023,54(21):7176-7185.
[12]孙仁爽,隋艳艳,闫红,等.基于指纹图谱结合一测多评法评价不同产地牻牛儿苗质量[J].中草药,2023,54(21):7186-7192.
SUN Renshuang,SUI Yanyan,YAN Hong,et al.Application of fingerprint combined with quantitative analysis of multi-components with a single-marker in quality evaluation of Erodium stephanianum[J].Chinese Traditional and Herbal Drugs,2023,54(21):7186-7192.
[13]田方,李军山,常云凤,等.一测多评法同时测定车前子配方颗粒中3种成分含量[J].河北工业科技,2018,35(6):459-464.
TIAN Fang,LI Junshan,CHANG Yunfeng,et al.Simultaneous determination of three constituents in Plantago Seed formula granules by QAMS[J].Hebei Journal of Industrial Science and Technology,2018,35(6):459-464.
[14]周倩,戴衍朋,王亮,等.HPLC法测定生甘草、炙甘草中6种成分[J].中成药,2016,38(2):378-382.
ZHOU Qian,DAI Yanpeng,WANG Liang,et al.Determination of six constituents in raw and baked Radix glycyrrhizae by HPLC[J].Chinese Traditional Patent Medicine,2016,38(2):378-382.
[15]关皎,王强,孙宇慧,等.UFLC法同时测定甘草及其炮制品种2种活性成分的含量[J].吉林医药学院学报,2018,39(4):241-243.
GUAN Jiao,WANG Qiang,SUN Yuhui,et al.Simultaneous determination of two components in Glycyrrhiza uralensis Fisch. and its processed products by UFLC[J].Journal of Jilin Medical University,2018,39(4):241-243.
[16]王晓亚,杜微波,马智玲,等.浮小麦配方颗粒特征图谱及4种核苷类成分定量研究[J].河北工业科技,2024,41(1):53-62.
WANG Xiaoya,DU Weibo,MA Zhiling,et al.Characteristic chromatogram of Fructus Tritici Levis formula granules and quantitative study of four nucleosides[J].Hebei Journal of Industrial Science and Technology,2024,41(1):53-62.
[17]田伟,李葆林,张闯,等.HPLC法同时测定薄荷配方颗粒中6种成分的含量[J].河北工业科技,2021,38(3):185-191.
TIAN Wei,LI Baolin,ZHANG Chuang,et al.Simultaneous determination of six constituents in Herba Menthae formula granules by HPLC[J].Hebei Journal of Industrial Science and Technology,2021,38(3):185-191.
