
糯玉米茎秆穿刺强度QTL分析与基因组选择
2024-09-26 00:00:00章慧敏张舒钰宋旭东张振良陆虎华陈国清郝德荣冒宇翔石明亮薛林周广飞
江苏农业学报 2024年7期

摘要:茎秆穿刺强度是衡量玉米茎秆机械强度和抗倒伏能力的重要指标之一,本研究以衍生于糯玉米自交系衡白522和通系5的198个重组自交系为试验材料,对茎秆穿刺强度进行数量性状位点(QTL)分析和基因组选择研究。单个环境QTL分析共检测到4个控制糯玉米茎秆穿刺强度的QTL,每个QTL的表型变异贡献率均小于10.00%,且仅在单个环境中被检测到;多个环境QTL分析共检测到8个QTL与环境互作,其加性效应总共可解释24.64%的表型变异,加性效应与环境互作贡献率为17.51%;上位性QTL分析共检测到4对QTL与QTL互作,可解释8.25%的表型变异。基因组选择中,当训练群体占群体总数的80%,随机选择500个标记即可获得较高的预测准确性;但是根据单个环境QTL分析结果,选择机率常用对数值排名前200的标记,即可大幅度提高基因组选择预测准确性。
关键词:糯玉米;茎秆穿刺强度;数量性状位点;基因组选择
中图分类号:S513文献标识码:A文章编号:1000-4440(2024)07-1191-08QTL analysis and genomic selection of rind penetrometer resistance in waxy maizeZHANG Huimin ZHANG Shuyu SONG Xudong ZHANG Zhenliang LU Huhua CHEN Guoqing HAO Derong MAO Yuxiang SHI Mingliang XUE Lin ZHOU Guangfei
(1.Jiangsu Yanjiang Institute of Agricultural Science, Nantong 226012, China;2.Jiangsu Collaborative Innovation Centre for Modern Crop Production, Nanjing 210095, China)
Abstract:Rind penetrometer resistance (RPR) is an important index that can be used to measure mechanical strength and lodging-resistance capability of maize stalk. In this study, a total of 198 recombinant inbred lines developed from waxy maize inbred lines Hengbai 522 and Tongxi 5 were used as test materials to perform quantitative trait locus (QTL) analysis and genomic selection (GS) study of RPR. Four QTLs for waxy maize PRP were detected by individual environmental QTL analysis, and contribution rate of phenotypic variation for each QTL was below 10.00% and was detected only in individual environment. Eight QTLs were detected to interact with the environment by multiple environmental QTL analysis, the additive effect could explain a total of 24.64% phenotypic variations, and the contribution rate of interaction between additive QTL and environment was 17.51%. Four pairs of QTL-QTL interactions were detected by epistatic QTL analysis, which could explain 8.25% of the phenotypic variation. In GS, when the training population size occupied 80% of the total population size, a relatively high prediction accuracy could be obtained by selecting 500 markers randomly. However, the genome selection prediction accuracy could be significantly improved by using top 200 markers for logarithm of odds values based on QTL analysis results of individual environmental.
Key words:waxy maize;rind penetrometer resistance;QTL(quantitative trait locus);genomic selection
倒伏是玉米生产中一种常见的现象,每年可造成5%~20%的产量损失[1]。引起玉米倒伏的原因有多种,外因主要包括强风、暴雨等恶劣天气以及不合理的田间管理和栽培措施等,内因主要是品种自身的茎秆机械强度。提高茎秆机械强度可以有效降低倒伏率,而玉米茎秆穿刺强度是衡量茎秆机械强度的重要指标之一[2-3]。
为了获得可用于玉米茎秆穿刺强度遗传改良的分子标记,国内外学者利用不同的遗传群体,鉴定到大量控制玉米茎秆穿刺强度的数量性状位点(QTL),也证实玉米茎秆穿刺强度遗传基础复杂,由微效多基因控制[4-5]。Flint-Garcia等[1]于2003年首次报道了玉米茎秆穿刺强度QTL的定位,该研究利用4个F2代群体,通过6个环境表型的鉴定,检测到36个茎秆穿刺强度QTL。Peiffer等[5]通过联合连锁和关联分析,鉴定到141个与玉米茎秆穿刺强度显著关联的遗传位点。Li等[6]利用2个重组自交系群体,在玉米第3染色体定位到1个茎秆穿刺强度主效QTL——qRPR3-1,通过单倍型分析,将qRPR3-1缩小至3.1 Mb的物理区间。Zhang等[7]利用多位点全基因组关联分析,检测到48个与玉米茎秆穿刺强度显著关联的遗传位点。Zhang等[8]利用图位克隆和关联分析,克隆了玉米茎秆强度基因stiff1,其启动子区有1 个27.2 kb的转座子插入,可降低该基因表达量,导致茎秆细胞壁纤维素和木质素含量提高,进而提高玉米茎秆穿刺强度和机械强度。Xu等[9]利用候选基因关联分析和突变体分析验证了ZmNR2调控玉米茎秆穿刺强度的生物学功能。
传统的分子标记辅助选择(MAS)依赖于QTL的准确性,且对效应值较大的QTL选择效果较好,不能有效选择微效的QTL[10]。得益于高通量测序技术的发展,Meuwissen等[11]于2001年提出了基因组选择(GS)的概念,即利用全基因组的标记估算育种群体中个体的育种值,根据育种值大小进行选择。与MAS不同的是,GS不需要检测显著的QTL,而是利用全基因组的标记进行选择,这样就可以估算出所有的遗传效应,解释全部的遗传变异,即便是标记微效,也能估算出来,可以大大提高选择效率[11]。Peiffer等[5]发现在不同的遗传群体中,对玉米茎秆穿刺强度进行GS,预测准确性存在显著差异。Liu等[12]的研究结果表明,将与玉米茎秆穿刺强度相关的位点作为固定效应加入GS数据模型,可显著提高预测准确性。
糯玉米是玉米传入中国之后,在栽培种植过程中发生变异而产生的一种新类型,与普通玉米相比,其籽粒中的淀粉几乎100%为支链淀粉,具有食用品质优良、营养价值丰富等特点,已成为老百姓菜篮子的重要食源[13]。抗倒性是糯玉米重要育种目标之一,然而上述研究均是以普通玉米为研究对象,对糯玉米茎秆穿刺强度的QTL分析和GS研究鲜有报道。本研究拟利用糯玉米骨干自交系衡白522×通系5衍生的重组自交系群体,在3个环境中鉴定群体的茎秆穿刺强度,检测控制糯玉米茎秆穿刺强度的QTL,分析QTL与环境互作效应及上位性效应,并探究GS在茎秆穿刺强度遗传改良中的应用潜力,以期为糯玉米抗倒品种的选育提供参考。
1材料与方法
1.1试验材料与基因型分析
试验材料为以衡白522和通系5为亲本,采用单籽粒传递法,连续自交7代构建的1个包含198个家系的重组自交系群体[14]。
利用Affymetrix microarray CGMB56K SNP Array基因芯片分析双亲和重组自交系群体的基因型,获得12 268个高质量且在双亲之间存在差异的单核苷酸多态性位点(SNP),将共分离的SNP标记视为一个bin,利用JoinMap4.0软件,将bin作为标记,利用Kosambi算法计算标记间的遗传距离,构建的遗传图谱包含2 703个bin,遗传图谱总长1 876.20 cM,平均遗传距离为0.73 cM[14]。
1.2田间试验与表型鉴定
田间试验分别于2017年(E1)、2018年(E2)和2019年(E3)在江苏沿江地区农业科学研究所如皋薛窑试验基地(32°23′N,120°33′E)进行,共3个环境。每个环境的田间试验均采用随机区组设计,2次重复,单行区,行长3.0 m,行距0.6 m,每行种植12株。
茎秆穿刺强度测定参考Flint-Garcia等[1]的方法,在玉米授粉14 d后,每行选取长势一致的玉米8株,利用浙江拓普仪器有限公司生产的植物茎秆强度测定仪YYD-1A,将横截面积为1.0 mm2的测头从茎秆的短轴方向垂直刺入地上部第三节间中部,读取并记录试验数据。
1.3表型数据分析
表型数据分析采用R 4.0.2软件(https:/www.r-project.org/)完成。利用psych包describe语言(https://CRAN.R-project.org/package=psych)计算群体的平均值、标准差、最小值、最大值、峰度和偏度等,使用corr.test语言计算不同环境之间的相关系数;利用stats包Shapiro.test语言(https://stat.ethz.ch/R-manual/R-patched/library/stats/)进行W测验;利用lme4包[15]lmer语言进行方差分析。
性状单环境广义遗传率计算参照公式H2=σ2/(σ2+σ2/r)×100%,多环境广义遗传率计算参照公式H2=σ2/(σ2+σ2/n+σ2/nr)×100%,其中σ2为基因型方差、σ2为基因型与环境互作方差、σ2为误差方差,n为环境数,r为重复数[16]。每个家系的最佳线性无偏预测值(BLUP)采用lme4包[15]中的lmer语言估算。
1.4QTL分析
QTL分析利用QTL IciMapping 4.1软件中的完备区间作图法(ICIM)[17]完成,在0.05显著性水平,通过1 000次排列,确定机率常用对数(LOD)值,其余参数为默认值。单个环境QTL分析利用BIP功能模块中的ICIM-ADD完成,用于检测控制茎秆穿刺强度的QTL;多个环境QTL分析利用MET功能模块中的ICIM-ADD完成,用于检测QTL与环境互作(QEI);上位性QTL分析利用MET功能模块中的ICIM-EPI完成,用于检测QTL与QTL之间的互作(QQI)。
1.5基因组选择
利用R 4.0.2软件中rrBLUP包构建rrBLUP模型[18]并进行GS,预测准确性定义为预测值与真实值之间的皮尔逊相关系数[19]。
1)在训练群体大小方面,设置不同的群体大小作为训练群体进行GS,分别选取群体中10%、20%、30%、40%、50%、60%、70%、80%和90%的材料作为训练群体,其余的材料作为测试群体,重复100次,标记数目为2 703个。
2)在标记密度方面,首先分别随机选择25个、50个、100个、200个、300个、400个、500个、600个、700个、800个、900个和1 000个标记进行GS,其次根据单环境QTL分析中每个标记的LOD值,从大到小排序,依次选取排名前25、50、100、200、300、400、500、600、700、800、900和1 000的标记,群体中80%的材料作为训练群体,其余20%的材料作为测试群体,重复100次。
2结果与分析
2.1群体表型分析
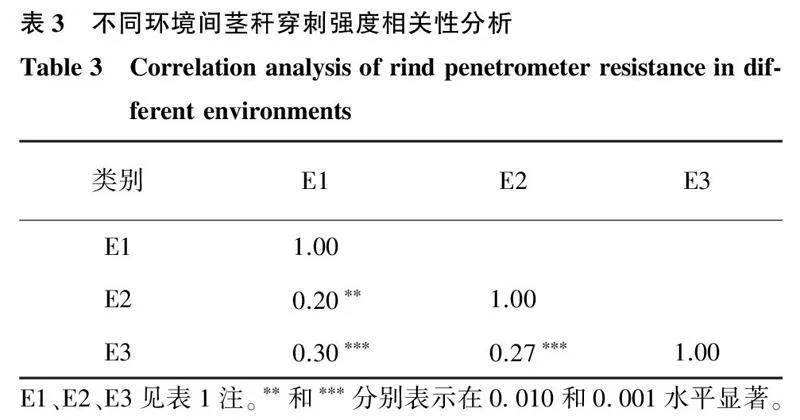
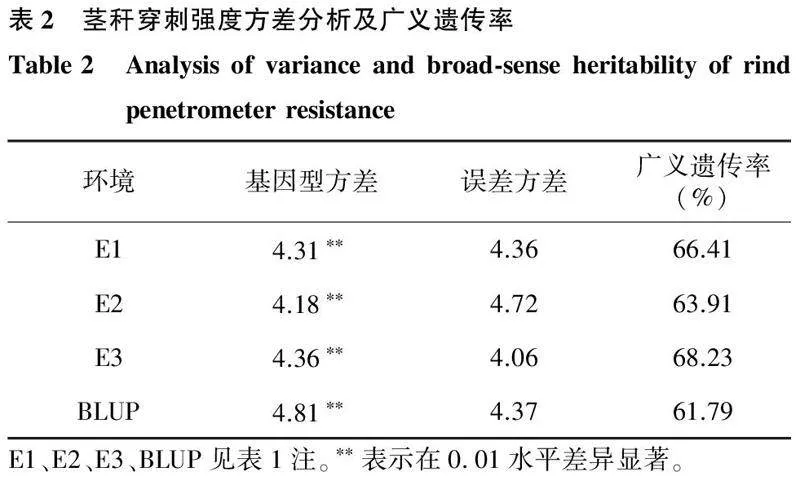
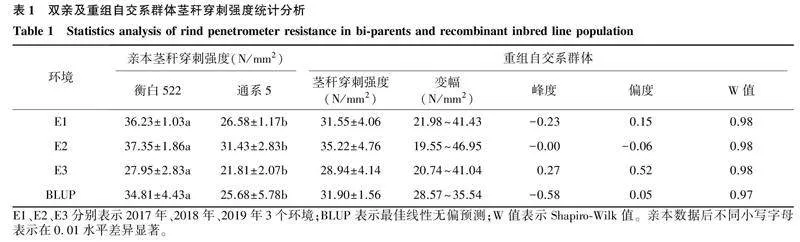
通过3个环境的表型鉴定,亲本衡白522的茎秆穿刺强度显著高于通系5的茎秆穿刺强度(表1)。不同环境下,茎秆穿刺强度具有较大的变异幅度(表1),不同基因型之间存在极显著差异(表2),表现出丰富的遗传变异。茎秆穿刺强度在群体中呈连续变异,偏度和峰度均介于-1.00~1.00,W值很大(P>0.05)(表1),符合正态分布,表现出数量性状特征,适合进行QTL定位。相关性分析结果表明,茎秆穿刺强度在各环境之间呈极显著正相关,但相关系数较小(表3),且环境方差(4.88)达到极显著水平,基因型与环境之间存在极显著的互作(6.74),表明茎秆穿刺强度易受环境影响。各环境中性状的广义遗传率较大,均高于60.00%(表2),表明基因加性方差是表型变异的主要因素。
2.2QTL分析结果
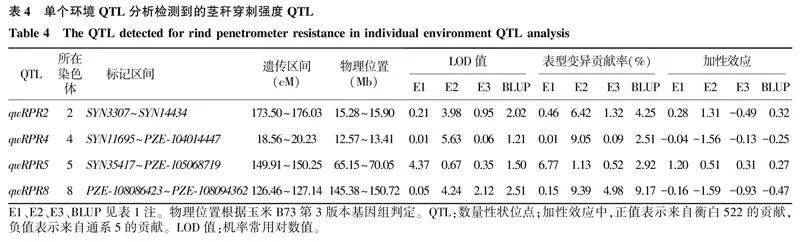
利用单个环境QTL分析,在LOD≥3.85的条件下,检测到4个控制糯玉米茎秆穿刺强度的QTL,分布在第2、第4、第5和第8染色体,共解释31.63%的表型变异贡献率(表4)。其中在E1环境下,检测到1个QTL(qwRPR5),在E2环境下检测到3个QTL(qwRPR2、qwRPR4和qwRPR8),在E3环境下和BLUP没有检测到QTL。
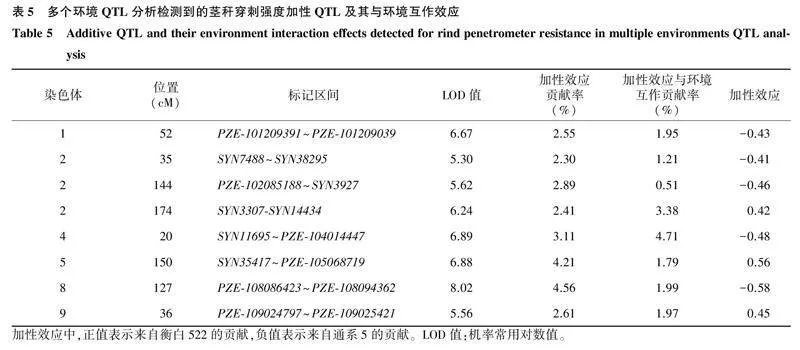
利用多个环境QTL分析,在LOD≥5.27的条件下,检测到8个QEI,分布在第1、第2、第4、第5、第8和第9染色体,加性效应总共可解释24.64%的表型变异,加性效应与环境互作贡献率为17.51%(表5)。其中单个环境QTL分析检测到的4个QTL均被检测到,说明这4个QTL与环境存在显著的互作效应。
利用上位性QTL分析,在LOD≥5.50的条件下,检测到4对QQI,位于不同的连锁群,包括8个遗传位点,可解释1.58%~2.80%的表型变异,总表型变异贡献率为8.25%(表6)。
2.3基因组选择结果
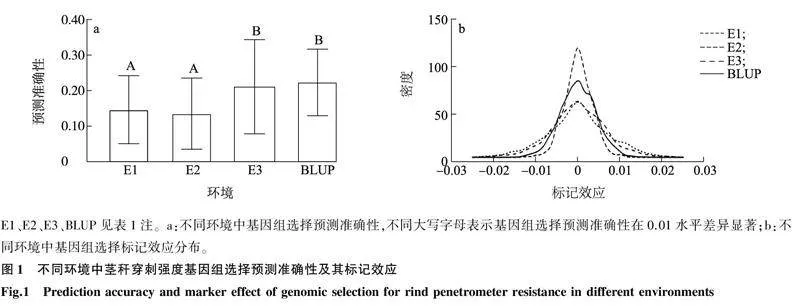
利用全基因组的2 703个标记,以群体中80%的材料作为训练群体,其余20%的材料作为测试群体,重复100次,E1、E2、E3和BLUP的基因组选择预测准确性分别为0.15、0.14、0.21和0.22(图1a),其标记效应呈正态分布(图1b)。不同环境中基因组选择预测准确性结果显示,E1和E2的基因组选择预测准确性之间没有显著差异,E3和BLUP的基因组选择预测准确性之间没有显著差异,E1、E2与E3、BLUP的基因组选择预测准确性之间存在显著差异(图1a),说明环境对GS预测准确性有一定的影响。尽管E3和BLUP在单环境QTL分析中没有检测到QTL,但其GS预测准确性显著高于E1和E2中的GS预测准确性(图1a),可能由于较严格的阈值,致使E3和BLUP中丢失的真实微效位点多于E1和E2中丢失的真实微效位点。利用3个环境的BLUP值作为表型值进行GS,预测准确性均高于利用单个环境的表型值进行GS,因此后续分析中均利用群体的BLUP值作为表型进行GS。
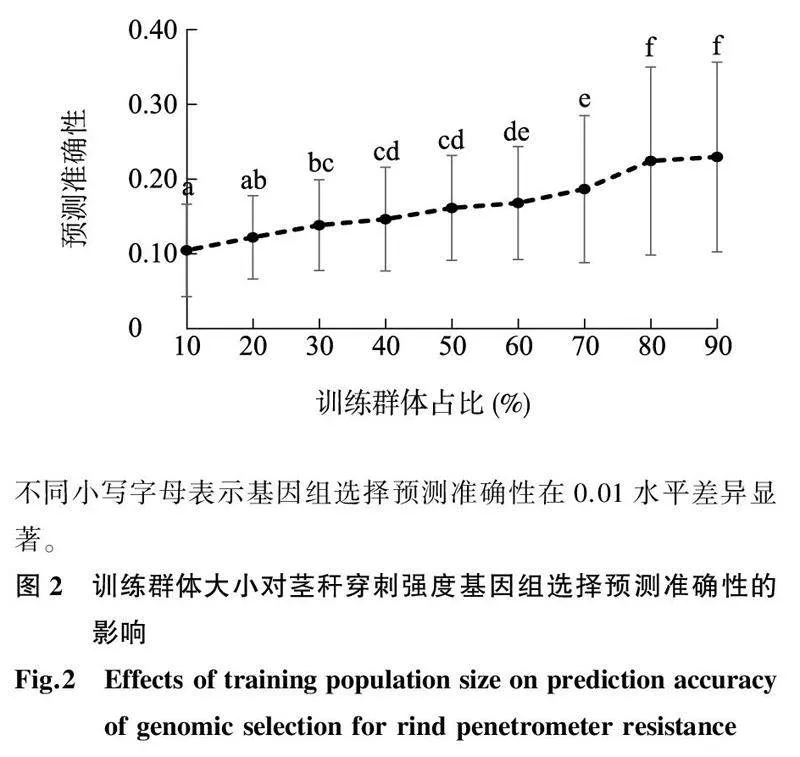
在训练群体大小方面,随着训练群体占比逐渐增大,GS预测准确性也逐渐提高。当训练群体数占群体总数的比例由10%增加到70%时,预测准确性缓慢提高,由0.10上升到0.18,当训练群体比例为80%时,预测准确性上升到0.22,当训练群体比例为90%时,预测准确性为0.23(图2)。
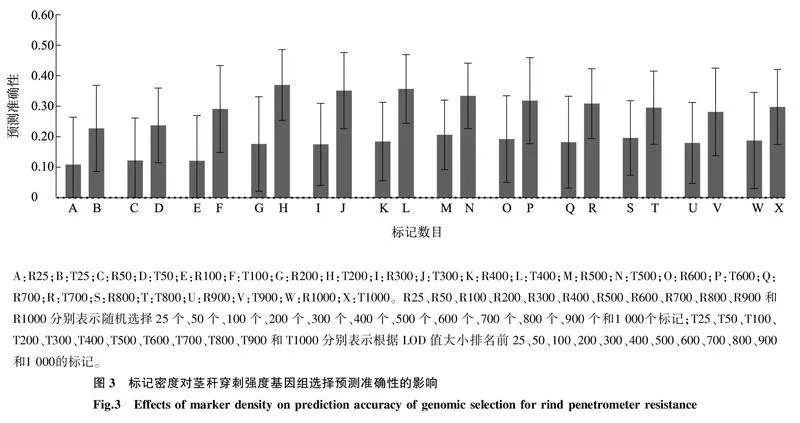
在标记密度方面,当随机选取的标记密度从25个增加到200个时,预测准确性快速提高,由0.11上升到0.18,当随机选择500个标记时,预测准确性为0.20,接近利用2 703个标记的预测准确性(0.22),之后随着标记密度的增加,预测准确性处于一个稳定水平(图3)。当根据LOD值大小选择标记时,预测准确性显著高于随机选择相同数目标记的预测准确性(图3)。当选择排名前200的标记时,预测准确性为0.37,与利用全基因组标记得到的结果相比大幅度提高,但之后随着标记密度的增加,预测准确性处于一个下降的趋势(图3)。
3讨论
本研究在单环境QTL分析中,仅在E1 和E2中分别检测到1个和3个控制糯玉米茎秆穿刺强度的QTL,而E3和BLUP中没有检测到QTL。第1个可能的原因是,较严格的阈值(3.85)造成了某些微效位点丢失[20]。当阈值取常用的2.50时,E1有2个QTL,E2有3个QTL,反而E3有6个QTL,BLUP有4个QTL。但如此低的阈值,会增加第一类错误发生的概率。因此,为检测到一定数量可靠的QTL,并降低假阳性率,尽管E3和BLUP中没有检测到QTL,本研究仍选择3.85作为阈值。与前期研究相比,本研究检测到的4个QTL均有报道。qwRPR2与Meng等[21]利用Zheng58×Chang7-2组配的双单倍体群体检测到的1个QTL位置重合,qwRPR4的物理位置与pQTL4-1相距0.5 Mb[12],qwRPR5区间内包含Peiffer等[5]检测到的8个SNP和Zhang等[7]检测到的2个SNP,且与pQTL5-1区间重合[12],qwRPR8与qRPR8区间重合[22]。
第2个可能的原因是,QTL与环境互作,即某一QTL仅在特定环境下表达,在其他环境下不表达,或在某些环境下表达强烈,而在其他环境下表达较弱,或在不同环境中的表现相反[23-24]。本研究利用多环境QTL分析,检测到8个QEI,其加性效应的表型变异总贡献率仅比加性效应与环境互作总贡献率高7.13个百分点。其中4个QEI与单环境QTL分析结果的位置重叠,而这4个QTL在单环境分析中仅在1个环境中被检测到,且有2个QTL(qRPR2和qRPR4)与环境互作贡献率大于其加性效应贡献率,表明尽管本群体中茎秆穿刺强度具有较高的广义遗传率,QTL与环境互作对表型变异也具有重要作用。
上位性效应是指非等位基因间相互作用引起的效应[25]。前期研究结果表明,上位性效应对玉米茎秆穿刺强度具有重要作用[1]。本研究利用上位性QTL分析,检测到4对QQI,共可解释8.25%的表型变异,仅约为加性效应表型变异贡献率的1/3(24.64%),说明本群体中上位性效应对表型变异的影响较小。
GS是随着现代测序技术发展而来的、一种利用覆盖全基因组高密度分子标记进行预测的育种方法,比较适合由微效多基因控制的复杂数量性状的遗传改良[11]。与MAS相比,GS不需要检测主效QTL,但其预测准确性受多种因素影响,其中包括群体大小和标记密度[26]。在研究群体大小对预测准确性的影响时,通常会考虑训练群体与测试群体规模比例。本研究发现,当训练群体的个体数目是测试群体的4.0倍时,预测准确性达到最大值。该结果与前人的研究结果有所不同,Guo等[27] 研究玉米籽粒锌含量时发现,当训练群体与测试群体大小一样时,预测准确性达到最大值,而Liu等[28]对玉米株型、产量等相关性状进行GS时,发现当训练群体大小是测试群体的3.0倍时,可实现最高水平的预测。这种情况可能是使用群体大小和目标性状不同导致的。
GS的基本原理是假设所有分子标记中至少有1个标记与所有控制目标性状的QTL/基因处于连锁不平衡状态[11],通过提高标记密度可以确保功能标记被充分覆盖,进而提高预测准确性。本研究结果表明,当随机选择500个标记时,预测准确性与利用全基因组标记得到的结果接近,这与利用双亲群体对玉米穗腐病[29]、雄穗相关性状[30]GS的结果相似。根据QTL分析结果,选择与茎秆强度相关的标记,可以显著提高预测准确性,尤其是选择排名前200的标记,预测准确性较利用全基因组标记得到的结果显著提高,但之后随着标记密度的增加,预测准确性不升反降。在玉米穗行数GS分析中也得到了相似的结果[31]。上述结果说明,一方面通过增加标记密度可以提高预测准确性,选择与茎秆强度相关的标记,可以在较少的标记密度下,实现较高水平的预测;另一方面若是标记数目太多,可能因为标记的随机效应,影响预测准确性。因此,利用QTL定位或全基因组关联分析结果,选择适当的标记数目,可以在提高预测准确性的同时,降低检测成本。
参考文献:
[1]FLINT-GARCIA S A, JAMPATONG C, DARRAH L L, et al. Quantitative trait locus analysis of stalk strength in four maize populations[J]. Crop Science,2003,43(1):13-22.
[2]KAMRAN M, CUI W, AHMAD I, et al. Effect of paclobutrazol, a potential growth regulator on stalk mechanical strength, lignin accumulation and its relation with lodging resistance of maize[J]. Plant Growth Regulation,2018,84(2):317-332.
[3]丰光,黄长玲,邢锦丰. 玉米抗倒伏的研究进展[J]. 作物杂志,2008(4):12-14.
[4]王夏青,宋伟,张如养,等. 玉米茎秆抗倒伏遗传的研究进展[J]. 中国农业科学,2021,54(11):2261-2272.
[5]PEIFFER J A, FLINT-GARCIA S A, DE LEON N, et al. The genetic architecture of maize stalk strength[J]. PLoS One,2013,8(6):e67066.
[6]LI K, YAN J B, LI J S, et al. Genetic architecture of rind penetrometer resistance in two maize recombinant inbred line populations[J]. BMC Plant Biology,2014,14:152.
[7]ZHANG Y L, LIU P, ZHANG X X, et al. Multi-locus genome-wide association study reveals the genetic architecture of stalk lodging resistance-related traits in maize[J]. Frontiers in Plant Science,2018,9:611.
[8]ZHANG Z H, ZHANG X, LIN Z L, et al. A large transposon insertion in the stiff1 promoter increases stalk strength in maize[J]. The Plant Cell,2020,32(1):152-165.
[9]XU S H, TANG X, ZHANG X M, et al. Genome-wide association study identifies novel candidate loci or genes affecting stalk strength in maize[J]. The Crop Journal,2023,11(1):220-227.
[10]NAKAYA A, ISOBE S N. Will genomic selection be a practical method for plant breeding?[J]. Annals of Botany,2012,110(6):1303-1316.
[11]MEUWISSEN T H E, HAYES B J, GODDARD M E. Prediction of total genetic value using genome-wide dense markers maps[J]. Genetics,2001,157:1819-1829.
[12]LIU X G, HU X J, LI K, et al. Genetic mapping and genomic selection for maize stalk strength[J]. BMC Plant Biology,2020,20(1):196.
[13]赵久然,卢柏山,史亚兴,等. 我国糯玉米育种及产业发展动态[J]. 玉米科学,2016,24(4):67-71.
[14]HAO D R, XUE L, YUAN J H, et al. Genetic dissection of starch paste viscosity characteristics in waxy maize revealed by high-density SNPs in a recombinant inbred line population[J]. Molecular Breeding,2017,37:50.
[15]BATES D, MCHLER M, BOLKER B, et al. Fitting linear mixed-effects models using lme4[J]. Journal of Statistical Software,2014,67:1-48.
[16]KNAPP S J, STROUP W W, ROSS W M. Exact confidence intervals for heritability on a progeny mean basis1[J]. Crop Science,1985,25:192-194.
[17]MENG L H, LI H, ZHANG L Y, et al. QTL icTGmpJJ255PdjGc0caTYcJQ==iMapping:integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations[J]. The Crop Journal,2015,3(3):269-283.
[18]ENDELMAN J B. Ridge regression and other kernels for genomic selection with R package rrBLUP[J]. The Plant Genome,2011,4(3):250-255.
[19]CROSSA J, PEREZ P, HICKEY J, et al. Genomic prediction in CIMMYT maize and wheat breeding programs[J]. Heredity (Edinb),2014,112(1):48-60.
[20]ZHOU G F, MAO Y X, XUE L, et al. Genetic dissection of husk number and length across multiple environments and fine-mapping of a major-effect QTL for husk number in maize (Zea mays L.)[J]. The Crop Journal,2020,8(6):1071-1080.
[21]MENG Y J, LI J H, LIU J J, et al. Ploidy effect and genetic architecture exploration of stalk traits using DH and its corresponding haploid populations in maize[J]. BMC Plant Biol,2016,16:50.
[22]HU H X, MENG Y J, WANG H W, et al. Identifying quantitative trait loci and determining closely related stalk traits for rind penetrometer resistance in a high-oil maize population[J]. Theoretical and Applied Genetics,2012,124(8):1439-1447.
[23]LI Z K, YU S B, LAFITTE H R, et al. QTL x environment interactions in rice. I. heading date and plant height[J]. Theoretical and Applied Genetics,2003,108(1):141-153.
[24]JANSEN R C, VAN OOIJEN J W, STAM P, et al. Genotype-by-environment interaction in genetic mapping of multiple quantitative trait loci[J]. Theoretical and Applied Genetics,1995,91:33-37.
[25]BATESON W. The progress of genetics since the rediscovery of Mendel’s papers[J]. Progress Rei Botanicae,1906,1:368.
[26]XU Y B, LIU X G, FU J J, et al. Enhancing genetic gain through genomic selection:from livestock to plants[J]. Plant Communication,2020,1(1):100005.
[27]GUO R, DHLIWAYO T, MAGETO E K, et al. Genomic prediction of kernel zinc concentration in multiple maize populations using genotyping-by-sequencing and repeat amplification sequencing markers[J]. Frontiers in Plant Science,2020,11:534.
[28]LIU X G, WANG H W, WANG H, et al. Factors affecting genomic selection revealed by empirical evidence in maize[J]. The Crop Journal,2018,6(4):341-352.
[29]周广飞,高夕全. 玉米禾谷镰孢菌穗腐病抗性基因组选择研究[J]. 江苏农业科学,2023,51(14):65-70.
[30]许加波,吴鹏昊,黄博文,等. 利用F2:3家系来源单倍体定位玉米雄穗相关性状QTL及全基因组选择[J]. 作物学报,2023,49(3):622-633.
[31]LIU L, DU Y F, HUO D A, et al. Genetic architecture of maize kernel row number and whole genome prediction[J]. Theoretical and Applied Genetics,2015,128(11):2243-2254.
(责任编辑:陈海霞)
