
磷酸化蛋白质的定量与化学计量分析方法的研究进展
2024-09-02 00:00:00刘媛翟睿吴帆楚占营赵洋戴新华方向俞晓平
分析化学 2024年5期



摘要 蛋白质翻译后修饰是赋予蛋白质生理功能的关键机制,其中可逆的磷酸化修饰在众多生命活动中起着开/关的作用,其异常变化通常与多种重大疾病过程密切相关。近年来,借助蛋白质组学技术和方法,磷酸化蛋白质的高通量、高精度定性与定量分析方法也得到了快速发展。本文综述了近年来基于“自下而上”策略的磷酸化蛋白质定量与化学计量分析方法的研究进展,包括磷酸化肽的富集方法、磷酸化肽质谱碎裂方式、定量分析方法以及磷酸化位点的化学计量分析方法,并对磷酸化蛋白质研究的发展趋势进行了讨论。
关键词 磷酸化蛋白质;富集方法;质谱;定量方法;化学计量;评述
蛋白质翻译后修饰(Post-translational modifications, PTMs)是赋予蛋白质生理功能的关键机制,包括磷酸化、糖基化和泛素化乙酰化等[1]。PTMs 在信号转导、细胞周期、发育与分化、细胞凋亡以及多种代谢通路调控中发挥了极为重要的作用[2-3]。经磷酸化修饰的蛋白质在催化、调节及蛋白相互作用等方面发挥着重要作用,是很多生命过程的关键调控“开关”[4],许多重大疾病(如心血管疾病、癌症、神经退行性疾病等)已被证明与一些蛋白质的磷酸化水平变化密切相关。因此,对磷酸化蛋白质的定量与化学计量分析对理解生命体代谢过程、疾病发生机制以及个体预防治疗等均具有重要意义[5]。
质谱技术具有高灵敏度、高精密度和高通量等优势,已被广泛用于磷酸化蛋白质的定量分析。然而,由于磷酸化蛋白质的丰度极低,并且易受到生物基质中高丰度肽段信号的干扰,采用质谱技术很难对磷酸化肽直接进行检测[6]。因此,对磷酸化肽进行高效、高特异性富集是实现对其准确定性和定量分析的必要条件。针对上述问题,研究者开发了多种针对磷酸化肽富集和定量分析的方法,以提高磷酸化肽的质谱检测灵敏度和定量分析的准确性,在此基础上,开发了不同的质谱分析技术和基于“自下而上”
策略的磷酸化蛋白质定量和化学计量分析方法。本文介绍了磷酸化肽常用的富集方法、碎裂方式以及定量方法,总结了磷酸化位点的化学计量学及其研究方法近年来的研究进展,并讨论了其未来的发展趋势。
1 磷酸化肽的分离富集方法
目前,对于磷酸化蛋白质的定量和化学计量分析多采用“自下而上”的研究策略,首先将蛋白质酶解成肽段,再利用质谱技术对这些肽段进行分析,实现磷酸化蛋白质的鉴定和研究。对磷酸化肽段进行高效、高特异性的富集分离是实现高精度定性和定量分析的关键步骤[7]。研究者已基于磷酸化肽段化学特性开发了不同富集方法,大体可分为金属离子亲和色谱法、离子交换色谱法和免疫亲和富集法(作用于pTyr 结构域)等[8]。
固定化金属亲和色谱法(Immobilized metal-ion affinity chromatography, IMAC)[9]和金属氧化物亲和色谱法(Metal oxide affinity chromatography, MOAC)[10]是两种最普遍的富集pSer、pThr 和pTyr 的亲和色谱技术[11]。IMAC 方法利用螯合作用将金属阳离子(Fe3+、Ga3+和Zr4+等)固定在磁珠、二氧化硅或树脂等基质上,再通过与带负电荷磷酸基团的亲和作用实现对磷酸化肽的选择性保留。MOAC 主要是利用两性金属氧化物(如TiO2、ZrO2 和Fe3O4 等)[12]和磷酸根基团的结合作用富集磷酸化肽。Jensen 等[13]发现IMAC 更适宜多磷酸化肽富集而MOAC 更适宜单磷酸化肽富集。Thingholm 等[14]基于这两个特点,将这两种技术联用,有效增加了磷酸化肽鉴定覆盖率。
离子交换色谱技术是基于带负电荷的磷酸化肽与色谱基质上的离子交换基团发生静电相互作用,主要有强阳离子交换法(Strong cation exchange, SCX)和强阴离子交换法(Strong anion exchange, SAX)。Quan 等[15]发现SCX 主要是吸附一些碱性磷酸化肽段(pIgt;8.0),而SAX 主要是吸附一些酸性磷酸化肽段(pI 介于2.91~6.45 之间)和一些强酸性磷酸化肽段(pIlt;3.0)。免疫亲和富集方法基于抗原-抗体反应,其特异性更强,但由于抗体开发周期长、价格昂贵以及结果重现性不佳等问题,主要用于富集丰度较低的酪氨酸磷酸化肽(pTyr)。SH2 结构域是已知最大的一类pTyr 识别结构域[16]。Bian 等[17]使用SH2 超亲体并结合Ti4+-IMAC,实现了pTyr 的高特异性富集,从9 个人类细胞系中鉴定了约20000 个不同的pTyr和1000 余个酪氨酸磷酸化位点,其中36%的位点为首次鉴定。
N-磷酸化发生在碱性氨基酸上,形成磷酰胺键(P—N)。针对N-磷酸化肽的富集,已开发出pHis 抗体[18]和pArg 抗体[19]用于实现pHis 和pArg 肽段的有效富集。此外,基于IMAC 材料亲和色谱保留时间差异的方法也被用于pHis 和pLys 肽段的有效富集[20-21]。为了提高N-磷酸化肽的覆盖率,研究者又开发了多种富集N-磷酸化肽的方法。如Hu[22]等制备了具有核壳结构的亚二微米硅球,在硅球表面键合双二甲基吡啶胺双锌分子,在中性条件下基于该材料的On-tip 富集方法实现了N-磷酸化肽段的高效、高选择性和快速富集,结合液相色谱-质谱联用分析技术,从HeLa 细胞中鉴定到3384 个N-磷酸化位点。然而, P—N 磷酰胺键具有较高的吉布斯自由能,在富集过程中易发生水解,因此,对于N-磷酸化肽的高效富集和检测仍是一项具有挑战性的研究工作。
2 磷酸化肽的质谱碎裂方式
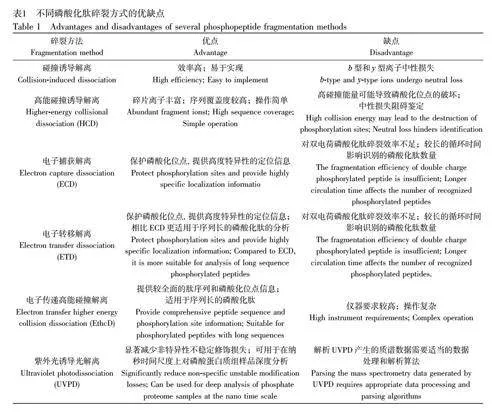
磷酸化肽离子的二级碎裂解离和解析是“自下而上”的磷酸化蛋白鉴定与定量分析策略的关键,因此磷酸化肽离子的碎裂方式在分析检测磷酸化位点过程中至关重要[23]。常用的磷酸化肽离子质谱碎裂方式包括碰撞诱导解离(Collision-induced dissociation, CID)、高能碰撞诱导解离(Higher-energy collisionaldissociation, HCD)、电子捕获解离(Electron capture dissociation, ECD)、电子转移解离(Electrontransfer dissociation, ETD)和紫外光诱导光解离(Ultraviolet photodissociation, UVPD)等。
CID 和HCD 是质谱实验中常用的磷酸化肽碎裂方法,均通过碰撞裂解肽段生成碎片离子,以获得关于肽段的结构和序列信息。CID 是通过气相肽离子与惰性气体(例如氦、氮)碰撞增加肽离子的内能,进而使肽离子碎裂[24-25]。虽然CID 的效率高,易于实现,但是CID 碎裂会造成磷酸化肽母离子及其b 型和y 型离子的非序列性中性丢失,丢失的中性部分可能是磷酸基团,影响磷酸化状态和结构的分析。HCD 是通过高能碰撞诱导解离产生碎片离子,常用于离子阱式质谱仪(如Orbitrap),利用多极碰撞单元或碰撞池实现, HCD 碰撞的能量比CID 强很多,因此会产生更多的碎片离子[26],与CID 相比, HCD 碎裂中b 型和y 型离子仍然会经历中性丢失,尽管中性丢失发生的频率或程度较小,但仍会阻碍鉴定和位点定位[27]。
ECD 和ETD 都是利用电子与磷酸化肽离子的相互作用实现碎裂。ECD 依赖于多电荷气相阳离子对低能电子的捕获,通过低能量的自由电子与质子化的多电荷蛋白质或肽离子相互作用放热而瞬间发生碎裂,主要产生由N—C 键断裂形成的c 和z 类型碎片离子[28]。Kruger 等[29]发现CID 能产生更多的碎片离子,而ECD 能解离不同类型的化学键,将ECD 与CID 联用可提高磷酸化蛋白测序的效率与准确度,增加磷酸化肽段鉴定覆盖率。ETD 利用低电子亲和力的阴离子将电子转移到多电荷肽阳离子,使其发生裂解,通过离子阱可有效地捕获阴离子试剂和阳离子肽[30]。主要产生由N—C 键α 断裂形成的c 和z 类型碎片离子,可以有效保留肽段骨架信息和磷酸化修饰信息,同时克服了ECD 中热电子传递和转移时间长的缺点。ECD 和ETD 在碎裂磷酸化肽中主要存在两个问题:(1)ECD 和ETD 的碎裂效率均取决于母离子的电荷状态,对于三电荷及更高电荷的肽都表现出良好碎裂效率,但对于最常见的双电荷磷酸化肽的碎裂效率不足;(2)ECD 和ETD 碎裂频率较低,需要较长的反应时间才能实现母离子解离,相对于碰撞碎裂的方法需要更长的循环时间,对鉴定磷酸化肽的数量有影响[31]。
EThcD 融合了ETD 和高能碰撞解离HCD 两种技术,其原理是先将较低能量的电子转移解离,保留更多的修饰位点信息,然后通过高能碰撞解离产生更多的碎片离子,提供更详细的序列信息,目前被广泛应用于磷酸化蛋白质组学[32-33]。虽然EThcD 鉴定的磷酸化肽数目少于HCD,但其序列覆盖率和位点定位的置信度比ETD 和HCD 显著增加。磷酸化肽段经ETD、HCD 和EThcD 碎裂后的MS/MS 谱图见图1,经ETD 碎裂后磷酸化肽段序列覆盖率较低,经HCD 碎裂产生足够多的b 离子和y 离子,但伴随着大量中性损失;经EThcD碎裂后的光谱图提供了足够的序列覆盖,且没有显著的修饰中性损失[34]。Penkert等[34]使用含有磷酸化的合成肽验证了EThcD 技术碎裂双电荷磷酸化肽的能力,证明EThcD 在双电荷磷酸肽的碎裂过程中优于ETD,并认为EThcD 是鉴定蛋白质中不稳定的磷酸化新位点的首选方法。
UVPD 是利用紫外光激发磷酸化肽内的键,引发磷酸化肽的离子解离,产生碎片离子,通过质谱对离子质荷比进行分析得到磷酸化位点和序列信息[35]。与HCD 相比, UVPD 产生的修饰损失显著减少(如单、双、三和四磷酸化肽离子分别减少47%、40%、50%和58%), b 离子和y 离子损失也较少,并且显示的离子得分显著增加,如图2 所示[36]。相比HCD, UVPD 提供的谱图信息更多,能显著减少磷酸化修饰基团的损失,并实现在纳秒时间尺度内深度分析磷酸化蛋白质组样品[36]。Robinson 等[37]发现,与HCD 相比, UVPD 提供了更多类型的子离子,并改善了子离子上的磷酸基团信息的保留,虽然阴性模式UVPD 在检测和测序高酸性磷酸化肽方面更优,但由于阴性模式下的电离效率较低,在进行大规模分析时可能无法充分覆盖整个样品的范围,因此, UVPD 适于与其它质谱技术结合(如HCD),充分利用各自的优势,提高磷酸化蛋白质的检测和测序能力,从而获得更可靠的结果。不同磷酸化肽碎裂方式的优缺点见表1。
3 磷酸化蛋白质的定量分析方法
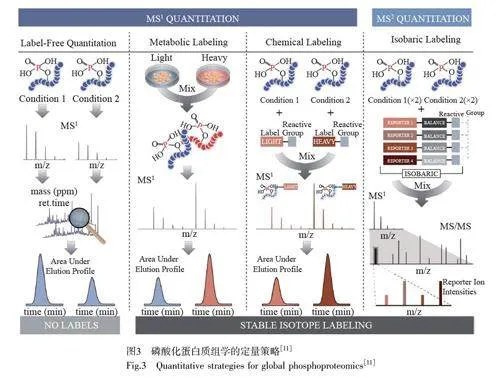
磷酸化蛋白质的定量分析主要采用“自下而上”的研究策略。相较于蛋白质组,磷酸化蛋白质组的定量分析更困难,具体表现在:(1)生物样品中非磷酸化肽含量较多,干扰磷酸化肽的质谱信号;(2)由于磷酸化肽段具有负电性,在质谱正离子模式下较难带正电荷,离子化效率低;(3)磷酰键比肽键更易断裂,增加了磷酸化肽定量的难度[38]。磷酸化肽段的定量分析方法有很多种,根据所用的质谱信息可分为一级质谱定量(MS1)和二级质谱定量(MS2),如图3 所示[11]。根据前处理过程的不同, MS1 定量又可分为非标记定量(Label-free)、代谢标记定量(Metabolic labeling)和化学标记定量(Chemical labeling)等;MS2定量主要是等重标记定量法。
3.1 非标记定量方法
Label-free 方法的准确性主要取决于液相色谱-质谱联用系统的稳定性。Label-free 主要有两种方法:其一为谱图计数法,对获得的目标肽段二级质谱图数量进行计数和比较,以此作为蛋白质表达量差异的定量依据;其二为离子强度法,利用测定的多肽母离子的色谱峰面积实现对蛋白质的表达量的定量分析[39]。与标记法相比, Label-free 无需使用特殊介质或昂贵的试剂,避免了在标记过程带来的实验误差,同时不需要考虑标记效率[40]。但是, Label-free 数据处理过程复杂、对仪器性能要求高、不适用于低丰度蛋白质检测。目前,已有研究者将Label-free 应用于重大疾病相关磷酸化蛋白质的定量分析,如Liu 等[41]利用高分辨率质谱法和开放式搜索方法对胃癌患者的肿瘤及癌组织的磷酸化蛋白质进行Label-free 定量分析,共鉴定了832 个与胃癌相关的磷酸化位点,为寻找放射治疗耐药性的重要靶点奠定了基础。此外, Label-free 也应用在识别生物标志物方面,如De Oliveira 等[42]为识别胃腺癌患者血清生物标志物,使用Label-free 对胃腺癌患者血清和非癌症患者血清中的33 种肽进行了差异定量分析,其中,19 种肽在胃腺癌患者血清样品中表达上调, 14 种肽表达下调。该研究表明,蛋白酶MMP-7 是一种有前景的生物标志物。
3.2 代谢标记定量方法
代谢标记定量方法是通过细胞正常代谢使蛋白质带上同位素标签。典型的代谢标记定量方法为氨基酸稳定同位素标记法(Stable isotope labeling by amino acid in cell culture, SILAC)[43]。SILAC 将轻、中或重型稳定同位素标记的必需氨基酸(赖氨酸和精氨酸)加入细胞培养基,通过细胞的正常代谢,新合成的蛋白会带上稳定同位素标签,将各类型蛋白质等量混合后进行质谱分析,通过比较一级质谱图中同位素峰形的面积而进行相对定量分析[44]。SILAC 的主要优势是在实验样品前处理时进行标记,从而将平行样品处理过程中可能引起的误差降至最低。但是, SILAC 需要经过多次细胞分裂才能确保定量分析稳定,因此不适合定量分析初代细胞。由于SILAC 可用于分析细胞、组织和体液等,因而被广泛应用于表征不同的生物样本蛋白质磷酸化差异[45]。Stepath 等[46]评估了Label-free、SILAC 和TMT 方法对磷酸化位点量化方面的性能,发现SILAC 在磷酸化位点的量化方面表现出较高的精度,是分析细胞培养模型中细胞信号传导的首选方法。Erber 等[47]利用SILAC 技术研究海马神经元HT-22 细胞中急性和慢性缺铁引起的磷酸化信号通路的变化,发现超过10%的磷酸化位点的相对丰度变化超过2 倍,表明缺铁和缺氧显著影响神经元细胞中的磷酸化信号通路。一些SILAC 衍生方法也被应用于磷酸化蛋白质组学分析,如脉冲式稳定同位素标记技术(Pulsed SILAC, pSILAC)[48]、高级稳定同位素标记技术(Super-SILAC)[49]等。与SILAC 直接分析混合后的轻重标记的样品不同, Pulsed SILAC 技术是在细胞培养过程中将“轻”、“中”、“重”氨基酸交替加入到培养基中,并培养特定时间,以跟踪和定量分析蛋白质的动态变化[50]。Wu 等[51]基于Pulsed SILAC 开发出差异同位素标记技术(DeltaSILAC)新策略,将细胞在不同时间点进行SILAC 标记,比较不同时间点的蛋白质组成的变化,该方法可以直接考察磷酸位点对蛋白质寿命的影响,进一步分析磷酸化位点对蛋白质表达寿命的影响。
3.3 化学标记定量方法
化学标记定量方法是采用物理和化学性质相似的化学试剂对生物样品进行标记,再采用液相色谱-质谱联用系统对生物样品进行定量分析。近十年来,化学同位素标记受到越来越多的关注,并实现了对生物样品中内源性代谢物的准确定量分析[52]。磷酸化蛋白质组学中最常用的化学标记定量方法是稳定同位素二甲基标记定量分析方法(Stable-isotope dimethyl labeling),该方法具有良好的标记效率、简单的实验过程以及低廉的试剂成本,可用于分析多种类型样本。二甲基标记采用甲醛(CH2O)和氰代硼氢化钠(NaBH3CN)两种稳定的同位素试剂标记蛋白质/肽的N 端或赖氨酸的氨基基团,使蛋白质/肽标记上不同的标签[53],可在每个标记位点与非标记对应物间产生28 个质量单位值差,在每个对应标记同位素上产生4 个质量单位值差,并且不会产生任何可检测的副产物[54]。由于二甲基适用性强,因而被广泛应用于重大疾病的磷酸化组学定量分析。Liu 等[55]使用稳定同位素二甲基标记结合高分辨率质谱法,在5-Fu耐药细胞系Bel/5-Fu 中共确定了8272 种独特的蛋白质和22095 个具有高定位置信性的磷酸化位点,并确定了肝细胞癌中减弱化学耐药性的潜在靶点。
3.4 等重标记定量方法
等重标记定量方法的原理是基于报告区与平衡区具有相同数量的总重同位素,当报告离子断开后即可通过比较样品之间相对报告离子强度对样品进行定量分析,包括同位素标记相对和绝对定量分析(Isobaric Tags for relative and absolute quantitation, iTRAQ)[56]、串联质谱标签(Tandem mass tags, TMT)[57]、氘同位素标记定量分析(Deuterium (2H) isobaric aminereactive tag, DiART)以及组合等压质量标签(Combinatorial isobaric mass tags, CMTs)等。
3.4.1 同位素标记相对和绝对定量分析
同位素标记相对和绝对定量分析(iTRAQ)的原理是利用多种同位素试剂标记蛋白多肽N 末端或赖氨酸侧链基团,经高精度串联质谱分析实现蛋白质组定量分析。目前, iTRAQ 试剂有4-plex 和8-plex,可分别标记4 组或8 组样品,包含与氨基结合的反应基团、不同分子量的报告基团以及不同分子量的质量平衡基团,当不同的报告基团分别与对应的平衡基团结合后会达到相同的质量,但在MS/MS 碎裂后产生独特的报告离子,以此对多肽进行定量分析[58]。iTRAQ 标记无偏倚且扫描范围广,可用于丝氨酸、苏氨酸和酪氨酸位点上的磷酸化定量分析[59]。为了鉴定参与调节MAT-LyLu 细胞转移的关键生物标志物,Xu 等[60]分别用TTX-S VGSCs 的特异性抑制剂HNTX-Ⅲ和激活剂JZTX-Ⅰ处理细胞,然后进行了基于iTRAQ 的定量磷酸化蛋白质组学分析,与对照组相比,在HNTX-Ⅲ和JZTX-Ⅰ处理组中分别鉴定出554 种和1779 种独特的磷酸化蛋白,其中, 55 种和36 种磷酸化蛋白质被鉴定为差异表达的蛋白质。差异表达的磷酸化蛋白与前列腺肿瘤的迁移和侵袭显著相关,因此有望成为前列腺癌症精确药物的潜在生物标记物。
3.4.2 串联质谱标签
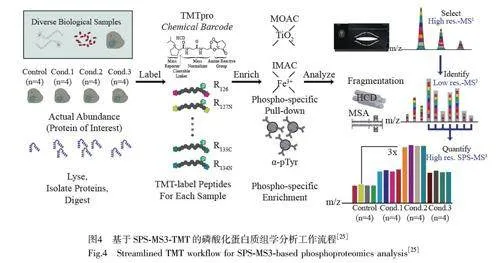
TMT 标记具有通量高、准确性高、对低丰度蛋白的鉴定效率高等优点,可同时对多个样品进行全蛋白质组定量分析,最新的16-Plex TMT 标记允许对多达16 个样本进行蛋白质组定量分析[61]。基于SPS-MS3-TMT(Synchronous precursor selection-MS3)的磷酸化蛋白质组学分析的主要流程如图4 所示[25]。TMT 标记定量磷酸化肽的方法已被广泛应用。Friedrich 等[62]使用TMT 标记,在非小细胞肺癌、亚型腺癌和鳞状细胞癌中定量分析8000 余种蛋白质,并确定了超过1400 个磷酸位点,为肺癌相关癌基因、肿瘤抑制因子及信号通路的蛋白质丰度和磷酸位点调节提供了相关定量信息。人参皂苷是一种重要的抗癌活性成分, Zou 等[63]利用TMT 标记定量分析人参皂苷治疗后乳腺癌MDA-MB-231 细胞中磷酸化蛋白质组的变化,在皂苷处理后的MDA-MB-231 细胞中鉴定出13 个位点的磷酸化状态发生变化,分析了人参皂苷的抗癌作用机制。TMT 标记反应通常在磷酸化肽富集之前,但TMT 标记对肽标记后会改变其化学性质,从而导致TiO2 富集磷酸肽的选择性发生变化, Ogata 等[64]设计了一种纳米级固相TMT 标记反应器,在磷酸化肽富集后进行TMT 标记,通过优化固相TMT 标记反应器中的pH 值、反相吸附剂和离子成功标记磷酸化肽,只需要少量TMT 试剂标记磷酸肽即可完成测定。此研究有助于纳米级TMT 标记应用于大规模磷酸蛋白质组学研究。
3.5 微量样品中磷酸化蛋白质的定量策略
磷酸化蛋白质定量组学分析通常采用数据依赖采集模式(Data dependent acquisition, DDA)采集质谱数据,在该模式下,需要有足够高丰度的磷酸肽离子才能实现有效的MS1 和MS2 分析。由于生物样本中磷酸化蛋白质丰度低,并且在正离子模式下质谱对磷酸化肽离子化效率较差,目前多需使用300~1000 μg 的提取蛋白才能实现可重复性的深度磷蛋白质组鉴定[65]。然而,临床样品通常只能提供少于10 μg 的提取肽,在如此微量的样品中实现磷酸化蛋白定量分析是一项重要且艰巨的任务。Budnik 等[66]利用等重标记法的MS1 信号强度取决于所有通道混合后的肽段总含量且各样品的MS2 定量离子信号互不干扰这一特性,开发了一种高通量SCoPE-MS(Single cell protEomics by mass spectrometry)方法,用于分析单细胞蛋白质组。选择TMT 标签的其中一个通道标记200 个细胞样品作为“Carrier”以提高MS1 信号强度,其余通道标记待测单细胞样品,所有通道混合后进行定量分析,在单细胞水平的蛋白质组学分析实验中,实现了对平均细胞直径仅为11 μm 的Jurkat 和U-937 细胞的蛋白质定量分析。上述的“Carrier”策略为微量样本中磷酸化蛋白质组的定量分析提供了新思路[67-69]。Yi 等[70]开发了一种BASIL(Boosting to amplify signal with isobaric labeling)等重标记策略,选择TMT 的一个通道标记“Carrier”样品,与其它通道混合后进行磷酸化肽富集和质谱检测,该设计显著增强了磷酸化肽的可检测性和可识别性,使可量化磷酸化位点的总数增加了4 倍以上。但是,在BASIL 策略中,研究样品和“Carrier”样品是在混合之后进行磷酸化肽富集和检测,每次分析都需要进行磷酸化肽富集处理,因而每个通道至少需要10 μg 的研究样品才能满足磷酸化肽分析的需求。Kwom 等[71]对上述策略进行了改进,使用富集后的高浓度磷酸化肽取代蛋白质组样品作为“Phosphocarrier”样品,该策略的原理示意图如图5 所示。来自“Phosphocarrier”样品的高浓度磷酸化肽贡献更强的MS1 信号强度(图5B),研究样品通道中的磷酸化肽无需进行富集处理且更易被选择用于MS2 分析(图5C),因而每个研究通道仅需低于1 μg 的样品即可实现全面的磷酸化蛋白质组分析。
酪氨酸磷酸化仅占总O-磷酸化蛋白质的0.05%,因此微量样品中酪氨酸的全面磷酸化蛋白质组学分析更困难。Chua 等[72]开发了BOOST(Broad-spectrum optimization of selective triggering)策略,利用酪氨酸磷酸酶抑制剂(Pervanadate, PV)处理的细胞作为TMT 标记的一个通道,选择性地增加了含磷酸化酪氨酸肽段的信号相对丰度,使磷酸化酪氨酸的定量深度提高6.3 倍,并且从1 mg T 细胞受体激活的Jurkat T 细胞中定量分析了2300 多种特异性酪氨酸磷酸化肽。
前处理样品转移过程中产生的非特异性表面吸附导致的样品损失也是微量样品磷酸化蛋白质定量分析面临的重要问题。Tsai 等[73]开发了一种基于串联Tip 的磷酸化蛋白质组学样品制备方法,该方法利用C18 与IMAC 结合形成串联C18-IMAC-C18,可快速、高效地富集磷酸化肽,并与SOP(Surfactant-assistedone-pot)[74]方法和iBASIL(Improved Boosting to amplify signal with isobaric labeling)[75]方法结合形成了一个简化的工作流程(图6),实现了高灵敏度、高通量的纳克级磷酸化蛋白质组定量分析,精确定量分析了100 个MCF10A 细胞中约600 条磷酸化肽和200 μm×200 μm×10 μm 的人类脾脏组织体素(相当于约100 个细胞)中约700 条磷酸化肽,为纳克级磷酸化蛋白质组分析开辟了新途径。
4 磷酸化位点的化学计量学
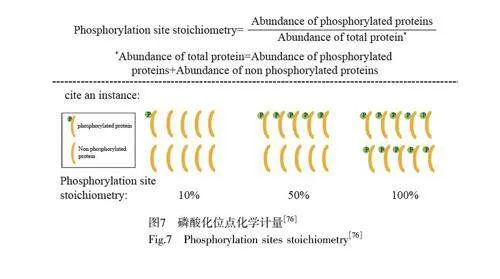
上述富集和定量方法帮助研究者鉴定到大量的磷酸化位点,并在一些研究中确定了磷酸化蛋白质的相对和绝对丰度。然而,蛋白的功能除了与特定位点的化学修饰有关外,还与该位点的修饰程度有关[76]。磷酸化位点占有率也称磷酸化位点化学计量,可通过测量给定位点的磷酸化形式相对于蛋白质总量的比例实现(图7)[76]。磷酸化位点的化学计量是磷酸化的重要属性,也是确定磷酸化位点生物学相关性的重要指标,对磷酸化依赖性的蛋白质调节过程研究具有重要意义[77]。与重大疾病相关的蛋白质磷酸化位点的化学计量研究对于深入理解疾病的发生机制以及开发新的治疗方法具有重要意义[78-79]。
4.1 绝对定量分析
由于蛋白质组的复杂性,在蛋白质组水平上确定特定位点的绝对磷酸化化学计量是一项具有挑战性的任务。早期,磷酸化化学计量均采用低通量方法,如定量蛋白质印迹法[80]。2003 年, Gerber 等[77]首次使用AQUA(Absolute quantification)法测量人体细胞周期依赖性的分离酶上Ser-1126 位点的磷酸化和非磷酸化形式的绝对量,从16 μg 样品中定量人类分离酶蛋白Ser-1126 位点的化学计量为34%。该方法的原理是向待测样品中加入已知量的稳定同位素标记磷酸化肽段和与其对应的稳定同位素标记非磷酸化肽段,通过比较同位素标记肽段与目标肽段的信号强度计算目标肽段的丰度。AQUA 被应用于精准量化不同样本的磷酸化位点化学计量,也可用于新的磷酸化位点化学计量定量方法的评估。但是, AQUA 在量化磷酸化位点化学计量时会受到肽电离效率的影响,并且AQUA 定量肽合成的成本较高。
4.2 非标记定量分析
采用同位素标记肽段进行定量分析成本较高, Steen 等[81]开发了一种非标记定量的方法,用于估计样品系列中磷酸化化学计量,当磷酸化肽及其对应的非磷酸化肽两种肽的信号强度变化具有相关性时,可通过监测磷酸化肽及其对应的非磷酸化肽的离子电流强度来测量磷酸化化学计量,基于此设计了相关计算方案:首先采用适当的归一化程序确定检测效率,然后通过监测磷酸化肽及其对应的非磷酸化肽之间的信号强度比即可得到目标磷酸化肽段的化学计量。该方法可用于定量分析多重磷酸化肽或蛋白酶切割效率受磷酸化位点影响的肽。该方法的局限是:定量分析的前提条件是目标磷酸肽及其对应的非磷酸化肽两种肽的信号强度变化必须相关;蛋白质的磷酸化比例必须达到一定程度(gt;10%),但不完全磷酸化。此外, Bekker-Jense 等[82]将多种实验条件信息集成到一个化学计量模型中,通过使用多通路复用量化磷酸化肽、非磷酸化肽和相应的蛋白质信号强度,然后在Perseus 插件中实现线性建模和化学计量计算,最终使用户能从Label-free 数据中计算磷酸化占用率。
4.3 磷酸化肽和非磷酸化肽的相对丰度分析
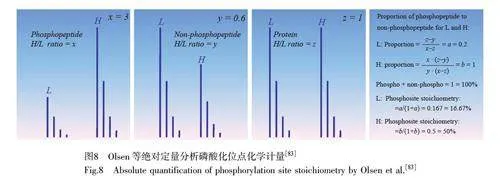
通过量化磷酸化肽和非磷酸化肽的相对丰度对磷酸化位点进行化学计量分析,通常是先量化样品中的磷酸化肽、非磷酸化肽和相应的蛋白质强度,再结合比例关系进行数学计算,进而得到位点的磷酸化水平。Olsen 等[83]假设磷酸化肽段与其对应的非磷酸化肽段比例达到1,从SILAC 轻标和SILAC 重标两种状态下计算磷酸化位点的绝对化学计量,具体计算公式如图8 所示,其中, x 表示SILAC 标记的轻、重磷酸化肽比例, y 表示SILAC 标记的轻、重非磷酸化肽比例, z 表示SILAC 标记的轻、重蛋白质比例,通过x、y、z 的SILAC 标记比率信息计算位点的磷酸化化学计量。采用该方法实现了磷酸化位点化学计量的大规模研究,并获得了人类培养细胞系HeLa S3 细胞周期中蛋白质和磷酸化位点动力学的系统视图。此外, Hogrebe 等[84]开发了一种基于三维多元回归模型(3DMM)的方法,使用量化方法中的磷酸化肽、非磷酸化肽和相应的蛋白质强度,结合肽的数量与肽的强度比例关系,在不同条件下对这3 种强度进行多元回归拟合,计算磷酸化程度。
近些年,已有大量通过磷酸酶去除磷酸化再结合同位素标记技术来测量蛋白质的磷酸化位点的研究[85-86]。Wu 等[87]将蛋白质裂解产物的两个相同等分试样分别进行虚拟处理以及磷酸酶处理,然后用稳定同位素进行差异化学标记。混合后,将获得的肽段序列与已知位点的数据库进行对比,基于重叠的非磷酸化形式的比率计算化学计量,最终确定了对数中期的酿酒酵母的5033 个磷酸化位点的化学计量。Chen 等[88]设计了一种二甲基标记结合磷酸酶去磷酸化的方法定量分析蛋白质中特定位点磷酸化水平,测得牛奶中α-S1 酪蛋白Ser130 位点和α-S2 酪蛋白Ser158 位点的磷酸化程度分别为97.2%和97.3%,这是首次在肽段水平上报道α-酪蛋白位点的特异性磷酸化程度。随后,研究者采用该方法测得未经过氧化叔丁醇溶液(Tert-butyl hydroperoxide, t-BHP)处理的Hsp27 上Ser82 的磷酸化程度为10.76%,经t-BHP 处理的Hsp27 上Ser82 的磷酸化程度为37.46%,表明该方法在分析蛋白质磷酸化动态变化方面具有可行性。
上述磷酸酶处理和同位素标记的方法具有工作流程简单、数据处理易于实现等优点,但仅适用于定量蛋白样品上有限数量位点的化学计量。在通过量化修饰肽和未修饰肽的相对丰度来进行化学计量测定的方法中,确保观察到的肽丰度变化具有统计学意义至关重要。为了使肽丰度变化更具有统计学意义, Lim 等[89]设计了贝叶斯模型(The Bayesian models),将化学计量建模为β 分布,可解决基于磷酸酶的磷酸化化学计量实验中的负化学计量的问题。
4.4 同位素标记非磷酸化肽段
使用磷酸化标准肽段定量磷酸化位点的化学计量会面临一些问题,其中合成定量用的磷酸化肽段中含有其对应的非磷酸化肽段,同时,磷酸化肽的低灵敏度也给质谱监测带来挑战。为规避上述问题,可通过磷酸酶处理或表达出无磷酸化修饰的QconCAT 蛋白,再通过归一化计算提高定量分析的准确性,最后只需利用同位素标记的非磷酸化肽段即可对目标肽段进行化学计量定量分析[90]。除此之外,Dominik 等[91]开发了一种基于磷酸酶处理和同位素标记用于测定复杂生物样品中的磷酸化化学计量的定量方法(Phosphatase-based phosphopeptide quantitation, PPQ)。该方法在酶切后加入同位素标记的非磷酸化肽段,然后将样品分为未处理和磷酸酶处理两部分,比较两部分样品的峰面积比即可确定磷酸化化学计量,因此,只需比较非磷酸化肽段的相对或绝对丰度即可确定单个肽的磷酸化化学计量。
4.5 磷酸化化学计量分析在激酶研究中的应用
激酶能够转移磷酸基团到特定的底物分子上,从而引发磷酸化修饰。磷酸化化学计量对于揭示激酶的调控机制和信号传导网络具有重要意义。激酶介导的磷酸化化学计量在细胞调控和疾病发展过程中具有重要作用。不同激酶对磷酸化位点的化学计量具有特异性,这种特异性可以调节底物的功能和细胞过程。此外,异常的磷酸化化学计量与许多疾病的发生发展密切相关,包括肿瘤、神经系统疾病和心血管疾病等。为了探究激酶对磷酸化的影响, Karayel 等[92]针对帕金森病患者中性粒细胞中Rab10-Thr73磷酸化水平进行了研究,发现帕金森病患者的富亮氨酸重复激酶2(Leucine-rich repeat kinase 2, LRRK2)突变会增加Rab10-Thr73 磷酸化水平。Tsai 等[93]采用两次富集结合磷酸酶处理的方法探究了不同激酶作用位点的磷酸化化学计量,在使用CK2、MAPK 和EGFR 作为肺癌症细胞中的基序靶向激酶的验证实验中,测量了1000 余个磷酸化位点,包括642 个CK2 靶向位点、940 个MAPK 靶向位点和366 个EGFR靶向位点,然后对不同激酶作用的磷酸化位点进行统计,发现CK2 有30%的磷酸化位点化学计量超过70%,而MAPK 和EGFR 只有少于15%的磷酸化位点化学计量大于70%,为激酶与磷酸化化学计量研究提供了新的研究思路。
5 总结和展望
磷酸化蛋白质参与多种生理和病理过程,具有重要的研究意义。不同类型的磷酸化肽段需采用不同的富集方法,质谱技术的发展也为磷酸化蛋白质研究提供了可靠的技术支撑。在此基础上的磷酸化位点化学计量学研究不仅对探究磷酸化位点和占有率具有生物学意义,也可用于分析磷酸化位点上游激酶的靶向位点和作用机制,这些成果可应用于重大疾病发病机制的探索和活性激酶抑制剂的临床研究,评估激酶抑制剂的靶向结合、剂量和疗效等,在生物医学上具有较大的应用潜力。此外,对微量样品进行全面的磷酸化蛋白质组学分析仍然是一项艰巨的任务,也是目前的研究热点,研究者采用不同的解决方案提高微量样品检测效率。磷酸化蛋白质的定量与化学计量分析方法在深入理解细胞信号传导、生物学过程和疾病机制方面发挥关键作用,未来的研究方向应主要集中在提高分析方法的精确度、灵敏度和通量,开发治疗重大疾病的靶向磷酸化位点的药物以及加速其在生物医学和精准医学领域的应用。
References
[1] DUAN G Y, WALTHER D. PLoS Comput. Biol. , 2015, 11(2): 23.
[2] CZUBA L C, HILLGREN K M, SWAAN P W. Pharmacol. Ther. , 2018, 192: 88-99.
[3] SEYREK K, LAVRIK I N. Apoptosis, 2019, 24(5-6): 385-394.
[4] GRIMSRUD P A, SWANEY D L, WENGER C D, BEAUCHENE N A, COON J J. ACS Chem. Biol. , 2010, 5(1): 105-119.
[5] THORNER J, HUNTER T, CANTLEY L C, SEVER R. Cold Spring Harbor. Perspect. Biol. , 2014, 6(12): a022913.
[6] MANN M, ONG S E, GRØNBORG M, STEEN H, JENSEN O N, PANDEY A. Trends Biotechnol. , 2002, 20(6): 261-268.
[7] CHAN C Y, GRITSENKO M A, SMITH R D, QIAN W J. Expert Rev. Proteomics, 2016, 13: 421-433.
[8] LOW T Y, MOHTAR M A, LEE P Y, OMAR N, ZHOU H, YE M. Mass Spectrom. Rev. , 2021, 40(4): 309-333.
[9] FICARRO S B, MCCLELAND M L, STUKENBERG P T, BURKE D J, ROSS M M, SHABANOWITZ J, HUNT D F,WHITE F M. Nat. Biotechnol. , 2002, 20(3): 301-305.
[10] LARSEN M R, THINGHOLM T E, JENSEN O N, ROEPSTORFF P, JØRGENSEN T J. Mol. Cell. Proteomics, 2005, 4(7):873-886.
[11] RILEY N M, COON J J. Anal. Chem. , 2016, 88(1): 74-94.
[12] KANSHIN E, MICHNICK S W, THIBAULT P. J. Proteome Res. , 2013, 12(6): 2905-2913.
[13] JENSEN S S, LARSEN M R. Rapid Commun. Mass Spectrom. , 2007, 21(22): 3635-3645.
[14] THINGHOLM T E, JENSEN O N, ROBINSON P J, LARSEN M R. Mol. Cell. Proteomics, 2008, 7(4): 661-671.
[15] QUAN Q, FENG J, LUI L T, SHI T, CHU I K. J. Chromatogr. A, 2017, 1498: 196-206.
[16] HUANG H, LI L, WU C, SCHIBLI D, COLWILL K, MA S, LI C, ROY P, HO K, SONGYANG Z, PAWSON T, GAO Y, LI SS. Mol. Cell. Proteomics, 2008, 7(4): 768-784.
[17] BIAN Y, LI L, DONG M, LIU X, KANEKO T, CHENG K, LIU H, VOSS C, CAO X, WANG Y, LITCHFIELD D, YE M, LIS S C, ZOU H. Nat. Chem. Biol. , 2016, 12(11): 959-966.
[18] MAKWANA M V, MUIMO R, JACKSON R F. Lab. Invest. , 2018, 98(3): 291-303.
[19] FUHRMANN J, SUBRAMANIAN V, THOMPSON P R. Angew. Chem. Int. Ed. , 2015, 54(49): 14715-14718.
[20] POTEL C M, LIN M H, HECK A J R, LEMEER S. Nat. Methods, 2018, 15(3): 187-190.
[21] HU Y, WENG Y, JIANG B, LI X, ZHANG X, ZHAO B, WU Q, LIANG Z, ZHANG L, ZHANG Y. Sci. China: Chem. ,2019, 62(6): 708-712.
[22] HU Y, JIANG B, WENG Y, SUI Z, ZHAO B, CHEN Y, LIU L, WU Q, LIANG Z, ZHANG L, ZHANG Y. Nat. Commun. ,2020, 11(1): 6226.
[23] SHI Wen-Hao, TONG Meng-Sha, LI Kai, WANG Yu-Shen, DING Chen. Prog. Biochem. Biophys. , 2018, 45(12): 1250-1258.
石文昊, 童梦莎, 李恺, 王钰珅, 丁琛. 生物化学与生物物理进展, 2018, 45(12): 1250-1258.
[24] WELLS J M, MCLUCKEY S A. Methods Enzymol. , 2005, 402: 148-185.
[25] PAULO J A, SCHWEPPE D K. Proteomics, 2021, 21(9): e2000140.
[26] ZHANG Y, FICARRO S B, LI S, MARTO J A. J. Am. Soc. Mass Spectrom. , 2009, 20(8): 1425-1434.
[27] CUI L, REID G E. Proteomics, 2013, 13(6): 964-973.
[28] ZUBAREV R A, KELLEHER N L, MCLAFFERTY F W. J. Am. Chem. Soc. , 1998, 120, 3265-3266.
[29] KRUGER N A, ZUBAREV R A, CARPENTER B K, KELLEHER N L, HORN D M, MCLAFFERTY F W. Int. J. Mass Spectrom. , 1999, 182-183: 1-5.
[30] SYKA J E P, COON J J, SCHROEDER M J, SHABANOWITZ J, HUNT D J. Proc. Natl. Acad. Sci. U. S. A. , 2004, 101:9528-9533.
[31] POTEL C M, LEMEER S, HECK A J R. Anal. Chem. , 2019, 91(1): 126-141.
[32] FRESE C K, ALTELAAR A M, TOORN H V D, NOLTING D, GRIEP-RAMING J, HECK A J, MOHAMMED S. Anal.Chem. , 2012, 84(22): 9668-9673.
[33] FRESE C K, ZHOU H, TAUS T, ALTELAAR A M, MECHTLER K, HECK A J, MOHAMMED S. J. Proteome Res. , 2013,12(3): 1520-1525.
[34] PENKERT M, HAUSER A, HARMEL R, FIEDLER D, HACKENBERGER C P R, KRAUSE E. J. Am. Soc. Mass Spectrom. , 2019, 30(9): 1578-1585.
[35] BRODBELT J S. Chem. Soc. Rev. , 2014, 43(8): 2757-2783.
[36] FORT K L, DYACHENKO A, POTEL C M, CORRADINI E, MARINO F, BARENDREGT A, MAKAROV A A,SCHELTEMA R A, HECK A J R. Anal. Chem. , 2016, 88(4): 2303-2310.
[37] ROBINSON M R, TALIAFERRO J M, DALBY K N, BRODBELT J S. J. Proteome Res. , 2016, 15(8): 2739-2748.
[38] SUI Shao-Hui, WANG Jing-Lan, CAI Yun, QIAN Xiao-Hong. Prog. Biochem. Biophys. , 2007, 34(3): 240-245.
隋少卉, 王京兰, 蔡耘, 钱小红. 生物化学与生物物理进展, 2007, 34(3): 240-245.
[39] MEGGER D A, BRACHT T, MEYER H E, SITEK B. Biochim. Biophys. Acta, 2013, 1834(8): 1581-1590.
[40] ANKNEY J A, MUNEER A, CHEN X. Annu. Rev. Anal. Chem. , 2018, 11(1): 49-77.
[41] LIU J, LI J, SUN Z, DUAN Y, WANG F, WEI G, YANG J H. J. Transl. Med. , 2021, 19(1): 339.
[42] DE OLIVEIRA T M, DE LACERDA J T J G, LEITE G G F, DIAS M, MENDES M A, KASSAB P, E SILVA C G S E,JULIANO M A, FORONES N M. Clin. Biochem. , 2020, 79: 61-69.
[43] MANN M. Nat. Rev. Mol. Cell Biol. 2006, 7(12): 952-958.
[44] HOEDT E, ZHANG G, NEUBERT T A. Adv. Exp. Med. Biol. , 2019, 1140: 531-539.
[45] CHEN X, WEI S, JI Y, GUO X, YANG F. Proteomics, 2015, 15(18): 3175-3192.
[46] SSTEPATH M, ZULCH B, MAGHNOUJ A, SCHORK K, TUREWICZ M, EISENACHER M, HAHN S, SITEK B, BRACHT T. J. Proteome Res. , 2020, 19(2): 926-937.
[47] ERBER L N, LUO A, GONG Y, BEESON M, TU M, TRAN P, CHEN Y. Nutrients, 2021, 13(1): 179.
[48] HAMMAREN H M, GEISSEN E M, POTEL C M, BECK M, SAVITSKI M M. Nat. Commun. , 2022, 13(1): 7431.
[49] LLUCENA A C R, AMORIM J C, DE PAULA LIMA C V, BATISTA M, KRIEGER M A, DE GODOY L M F, MARCHINI F K. Cell Stress Chaperones, 2019, 24(5): 927-936.
[50] BELLER N C, HUMMON A B. Mol. Omics, 2022, 18(7): 579-590.
[51] WU C, BA Q, LU D, LI W, SALOVSKA B, HOU P, MUELLER T, ROSENBERGER G, GAO E, DI Y, ZHOU H,FORNASIERO E F, LIU Y. Dev. Cell, 2021, 56(1): 111-124.e6.
[52] CHEN Y Q, SHEN H, YANG R J, WAN J B. Anal. Chim. Acta, 2021, 1179: 338839.
[53] BOERSEMA P J, AYE T T, VAN VEEN T A B, HECK A J R, MOHAMMED S. Proteomics, 2008, 8(22): 4624-4632.
[54] HSU J L, HUANG S Y, CHOW N H, CHEN S H. Anal. Chem. , 2003, 75(24): 6843-6852.
[55] LIU Z, WANG Y, YAO Y, FANG Z, MIAO Q R, YE M. J. Proteomics, 2019, 208: 103501.
[56] MERTINS P, UDESHI N D, CLAUSER K R, MANI D R, PATEL J, ONG S E, JAFFE J D, CARR S A. Mol. Cell.Proteomics, 2012, 11(6): M111. 014423.
[57] MCALISTER G C, HUTTLIN E L, HAAS W, TING L, JEDRYCHOWSKI M P, ROGERS J C, KUHN K, PIKE I, GROTHE R A, BLETHROW J D, GYGI S P. Anal. Chem. , 2012, 84(17): 7469-7478.
[58] TEDFORD N C, WHITE F M, RADDING J A. Briefings Funct. Genomics Proteomics, 2008, 7(5): 383-394.
[59] XIAO W, YANG Z, YAN X, FENG L, LONG L, TU T, DENG N, CHEN W, XIAO B, LONG H, ZENG Y. Front. Neurol. ,2020, 11: 626013.
[60] XU R, CHEN Y, WANG Z, ZHANG C, DONG X, YAN Y, WANG Y, ZENG Y, CHEN P. Toxins, 2021, 13(8): 554.
[61] NOTARAS M, LODHI A, BARRIO-ALONSO E, FOORD C, RODRICK T, JONES D, FANG H, GREENING D, COLAK D.Mol. Psychiatry, 2021, 26(12): 7760-7783.
[62] FRIEDRICH C, SCHALLENBERG S, KIRCHNER M, ZIEHM M, NIQUET S, HAJI M, BEIER C, NEUDECKER J, KLAUSCHEN F, MERTINS P. Nat. Commun. , 2021, 12(1): 3576.
[63] ZOU M, WANG J, GAO J, HAN H, FANG Y. Oncol. Lett. , 2018, 15(3): 2889-2898.
[64] OGATA K, TSAI C F, ISHIHAMA Y. J. Proteome Res. , 2021, 20(8): 4193-4202.
[65] NEEDHAM E J, HINGST J R, PARKER B L, MORRISON K R, YANG G, ONSLEV J, KRISTENSEN J M, HØJLUND K,LING N X Y, OAKHILL J S, RICHTER E A, KIENS B, PETERSEN J, PEHMØLLER C, JAMES D E, WOJTASZEWSKI JF P, HUMPHREY S J. Nat. Biotechnol. , 2022, 40(4): 576-584.
[66] BUDNIK B, LEVY E, HARMANGE G, SLAVOV N. Genome Biol. , 2018, 19(1): 161.
[67] TSAI C F, OGATA K, SUGIYAMA N, ISHIHAMA Y. Cell Rep. Methods, 2022, 2(1): 100138.
[68] CHUA X Y, SALOMON A. J. Proteome Res. , 2021, 20(6): 3330-3344.
[69] STOPFER L E, CONAGE-POUGH J E, WHITE F M. Mol. Cell. Proteomics, 2021, 20: 100104.
[70] YI L, TSAI C F, DIRICE E, SWENSEN A C, CHEN J, SHI T, GRITSENKO M A, CHU R K, PIEHOWSKI P D, SMITH R D, RODLAND K D, ATKINSON M A, MATHEWS C E, KULKARNI R N, LIU T, QIAN W J. Anal. Chem. , 2019, 91(9):5794-5801.
[71] KWON Y, LEE S, PARK N, JU S, SHIN S, YOO S, LEE H, LEE C. Anal. Chem. , 2022, 94(10): 4192-4200.
[72] CHUA X Y, MENSAH T, ABALLO T, MACKINTOSH S G, EDMONDSON R D, SALOMON A R. Mol. Cell. Proteomics,2020, 19(4): 730-743.
[73] TSAI C F, WANG Y T, HSU C C, KITATA R B, CHU R K, VELICKOVIC M, ZHAO R, WILLIAMS S M, CHRISLER W B,JORGENSEN M L, MOORE R J, ZHU Y, RODLAND K D, SMITH R D, WASSERFALL C H, SHI T, LIU T. Commun.Biol. , 2023, 6(1): 70.
[74] TSAI C F, ZHANG P, SCHOLTEN D, MARTIN K, WANG Y T, ZHAO R, CHRISLER W B, PATEL D B, DOU M, JIA Y,REDUZZI C, LIU X, MOORE R J, BURNUM-JOHNSON K E, LIN M H, HSU C C, JACOBS J M, KAGAN J,SRIVASTAVA S, RODLAND K D, STEVEN WILEY H, QIAN W J, SMITH R D, ZHU Y, CRISTOFANILLI M, LIU T,LIU H, SHI T. Commun. Biol. , 2021, 4(1): 265.
[75] TSAI C F, ZHAO R, WILLIAMS S M, MOORE R J, SCHULTZ K, CHRISLER W B, PASA-TOLIC L, RODLAND K D,SMITH R D, SHI T, ZHU Y, LIU T. Mol. Cell. Proteomics, 2020, 19(5): 828-838.
[76] PRUS G, HOEGL A, WEINERT B T, CHOUDHARY C. Trends Biochem. Sci. , 2019, 44(11): 943-960.
[77] GERBER S A, RUSH J, STEMMAN O, KIRSCHNER M W, GYGI S P. Proc. Natl. Acad. Sci. U. S. A. , 2003, 100(12):6940-6945.
[78] ALSUFAYAN T A, MYERS E J, QUADE B N, BRADY C T, MARSHALL A, HAQUE N, DUFFEY M E, PARKER M D.Int. J. Mol. Sci. , 2021, 22(23): 12817.
[79] FAN Y, NIRUJOGI R S, GARRIDO A, RUIZ-MARTINEZ J, BERGARECHE-YARZA A, MONDRAGON-REZOLA E,VINAGRE-ARAGON A, CROITORU I, GOROSTIDI PAGOLA A, PATERNAIN MARKINEZ L, ALCALAY R,HICKMAN R A, DURING J, GOMES S, PRATUSEVICIUTE N, PADMANABHAN S, VALLDEORIOLA F, PEREZ SISQUES L, MALAGELADA C, XIMELIS T, MOLINA PORCEL L, MARTI M J, TOLOSA E, ALESSI D R, SAMMLER E M. Acta Neuropathol. , 2021, 142(3): 475-494.
[80] COLYER J. Ann. N. Y. Acad. Sci. , 1998, 853: 79-91.
[81] STEEN H, JEBANATHIRAJAH J A, SPRINGER M, KIRSCHNER M W. Proc. Natl. Acad. Sci. U. S. A. , 2005, 102(11):3948-3953.
[82] BEKKER-JENSEN D B, BERNHARDT O M, HOGREBE A. Nat. Commun. , 2020, 11(1): 787.
[83] OLSEN J V, VERMEULEN M, SANTAMARIA A, KUMAR C, MILLER M L, JENSEN L J, GNAD F, COX J, JENSEN T S,NIGG E A, BRUNAK S, MANN M. Sci. Signal. , 2010, 3(104): ra3.
[84] HOGREBE A, VON STECHOW L, BEKKER-JENSEN D B, WEINERT B T, KELSTRUP C D, OLSEN J V. Nat. Commun. ,2018, 9(1): 1045.
[85] HEGEMAN A D, HARMS A C, SUSSMAN M R, BUNNER A E, HARPER J F. J. Am. Soc. Mass Spectrom. , 2004, 15(5):647-653.
[86] PFLIEGER D, JUNGER M A, MÜLLER M, RINNER O, LEE H, GEHRIG P M, GSTAIGER M, AEBERSOLD R. Mol.Cell. Proteomics, 2008, 7(2): 326-346.
[87] WU R, HAAS W, DEPHOURE N, HUTTLIN E L, ZHAI B, SOWA M E, GYGI S P. Nat. Methods, 2011, 8(8): 677-683.
[88] CHEN S H, LIN Y C, SHIH M K, WANG L F, LIU S S, HSU J L. Molecules, 2020, 25(22): 5316.
[89] LIM M Y, O’BRIEN J, PAULO J A, GUGI S P. J. Proteome Res. , 2017, 16(11): 4217-4226.
[90] JOHNSON H, EYERS C E, EYERS P A, BEYNON R J, GASKELL S J. J. Am. Soc. Mass Spectrom. , 2009, 20(12): 2211-2220.
[91] DOMANSKI D, MURPHY L C, BORCHERS C H. Anal. Chem. , 2010, 82(13): 5610-5620.
[92] KARAYEL O, TONELLI F, VIRREIRA WINTER S, GEYER P E, FAN Y, SAMMLER E M, ALESSI D R, STEGER M,MANN M. Mol. Cell. Proteomics, 2020, 19(9): 1546-1560.
[93] TSAI C F, KU W C, CHEN Y J, ISHIHAMA Y. Methods Mol. Biol. , 2017, 1636: 313-325.
猜你喜欢
食品安全导刊(2021年20期)2021-08-30 06:39:48
质谱学报(2019年5期)2019-09-24 02:18:32
企业技术开发·下旬刊(2016年11期)2016-12-27 10:32:34
中国科技博览(2016年21期)2016-11-14 10:49:04
现代经济信息(2016年24期)2016-11-09 13:33:23
商场现代化(2016年15期)2016-08-23 18:28:04
中国市场(2016年24期)2016-07-06 04:07:57
现代经济信息(2016年7期)2016-05-19 10:00:00
当代化工研究(2016年5期)2016-03-20 16:21:35
特产研究(2014年4期)2014-04-10 12:54:22
